Spinal Epidermoid Tumors: Case Report and Review of the Literature
Article information

Abstract
Spinal epidermoid tumors are rare, benign tumors that are either acquired from trauma, surgery, or lumbar puncture or arise as congenital lesions, particularly spinal dysraphisms. We report a case of a massive spinal epidermoid tumor and review the literature with a focus on the surgical outcomes. A 71-year-old female patient presented after a fall with subsequent symptoms of severe back and hip pain, as well as loss of motor strength in the left leg. Her magnetic resonance imaging demonstrated a T2/short tau inversion recovery hyperintense mass extending from the level of the T10–11 disc caudally through S2. A biopsy was recommended to determine whether the tumor was radio- or chemo-sensitive. The patient underwent a L4 laminectomy and a pearly-white tumor was encountered, with a subsequent biopsy confirming it to be an epidermoid tumor. The following conclusions can be drawn from a review of the literature. Spinal epidermoid tumors are more common in women and tend to present in younger patients (median age of 23). The majority of patients had acquired lesions (46%). In terms of surgical outcomes for adherent tumors, gross total resection was found to provide optimal outcomes, with 90% of patients improving clinically after surgery.
INTRODUCTION
Spinal epidermoid tumors are rare, comprising of less than 1% of tumors involving the spine [1]. These tumors arise from pathological displacement of epidermal cells into the spinal canal. Therefore, these tumors can be congenital, when there is improper closure of the neural tube; or acquired, in patients who have had prior lumbar punctures, trauma, or surgery [2,3]. Spinal epidermoids are typically found in the lumbosacral region but can be found in other locations as well [4]. They appear, intraoperatively, as white masses that are encapsulated and are commonly referred to as “pearly tumors.” [5] Their imaging and gross appearance at surgery is similar to those found in the cranium. Patients usually present with pain as well as neurologic dysfunction that may include: muscle weakness and atrophy, sensory disturbances, and loss of sphincter control [3,5].
METHODS
We present a case of a massive intraspinal epidermoid and review the literature; highlighting the clinical features, imaging, and treatment of these tumors. The extensive literature review was conducted using PubMed with search terms: “intraspinal epidermoid tumor,” “intraspinal epidermoid cyst,” “intradural spinal epidermoid tumor” and “intradural spinal epidermoid cyst.” The following information was collected: age, sex, symptoms, signs, size of the tumor, treatment, surgical results, outcome, follow-up, congenital or acquired, and tumor adhesion.
CASE REPORT
The patient is a 71-year-old female with a past medical history of hypertension who was admitted after a fall. The patient developed severe lower back and hip pain. She lost the ability to walk even with assistance. She reports having bilateral numbness in both legs that has been present since a spinal surgery over 3 decades ago, where she underwent lumbar surgery for decompression. The surgery was complicated by discovery of a spinal mass that upon attempted resection resulted in nerve root injury with permanent leg weakness and numbness. Since that time, the patient has lost her ability to urinate and defecate normally. In addition, she had a complete right foot drop.
The patient had 5/5 power on right hip flexion and knee extension, 0/5 power on right foot dorsiflexion and plantarflexion, 1/5 power in left hip flexion, knee extension, foot dorsiflexion and plantar flexion. Sensation was grossly intact to light touch and her reflexes were normal without hyperreflexia. Computed tomography (CT) scan of her pelvis demonstrated right alar sacral fracture extending to Dennis Zone II at the S1 level and a nondisplaced left lateral sacral alar fracture with lambda type configuration. There was also suggestion of a lumbar mass extending from L3 to S1 with significant bony remodeling of the spinal canal (Fig. 1A). Subsequent magnetic resonance imaging (MRI) of the thoracic and lumbar spine demonstrated a T2/short tau inversion recovery (STIR) hyperintense, tepidly enhancing intracanalicular expansive mass extending from the level of the T10–11 disc caudally through S2 (Fig. 1B). As seen on CT as well, there was chronic osseous remodeling and erosion of the L4 and L5 vertebral bodies and sacrum, with tumor protruding through the L4, L5, and sacral neural foramen.

(A) Sagittal computed tomography of the lumbar spine demonstrating osseous remodeling from L4 through the sacrum. (B) T2 sagittal magnetic resonance imaging demonstrating expansive mass located at T10–11 intervertebral disc through S2.
Based on the history and imaging findings; the differential diagnosis included myxopapillary ependymoma, ependymoma, schwannoma, dermoid tumor, and epidermoid tumor. Given her severe and chronic neurological deficits and extensive nature of the tumor with possible adhesion of the tumor to the nerve roots, an aggressive gross total resection (GTR) was avoided, as it would be unlikely to change her functional status. However, a biopsy was recommended in case the tumor was radioor chemo-responsive for palliation of her pain.
The patient underwent a partial L4 laminectomy for biopsy. The dura was displaced dorsally and was more superficial than in typical lumbar exposures. The thecal sac was opened and a pearly white substance was encountered. An epidermoid tumor was suspected based on this gross appearance (Fig. 2). Biopsy specimens were sent for pathological analysis. Electromyography (EMG) was used to ensure that none of the lumbosacral nerve rootlets were disturbed and there was no abnormal firing indicating irritation. Furthermore, no rootlets were seen under the microscope.

Appearance of epidermoid tumor during surgical biopsy. The classic pearly white tumor is seen extruding from the durotomy. Cr, cranial; Ca, caudal.
Pathology confirmed the diagnosis as epidermoid tumor with keratinizing squamous epithelium without adnexal structures or hair. At her 3-month follow-up, the patient reported significant improvement in pain but continued to have incontinence with no change in her chronic motor and sensory deficits. The pain improvement was attributed to partial healing of her sacral fractures, as opposed to the tumor biopsy. No further treatment for the tumor was initiated however, radiation is an option for palliative treatment in the future if surgical management is not feasible.
LITERATURE REVIEW
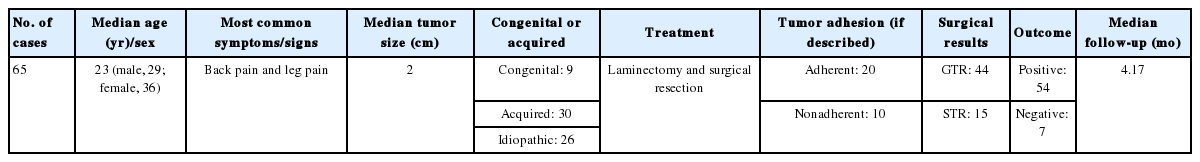
A total of 65 cases (including the current case) were reviewed in the literature (Supplementary Table 1) [1-29]. There were a total of 29 male patients and 36 female patients. The median age was 23 years (range, 0–71 years). The most common symptoms were back and leg pain. Thirty cases were acquired from either lumbar puncture or prior surgery, 9 cases were deemed congenital and 26 cases were idiopathic. The most common treatment was surgical; specifically, laminectomies and intradural tumor resection. A total of 44 patients had GTRs, 15 had subtotal resections (STRs), and in 8 patients the type of resection could not be determined. Two patients developed a recurrence of their tumors after surgery. One patient had a GTR first and then developed a recurrence, which was then partially resected in a subsequent surgery. Another patient had a STR and then developed a recurrence, which was then partially resected again. 26 patients (40%) had tumors that were described as adherent, 10 patients (15%) had tumors that were not adherent, and for the remaining cases tumor adherence could not be determined. The median follow-up time was 4.2 months amongst all of the reviewed studies. A summary of demographics and outcomes from the literature review is presented in Table 1.
DISCUSSION
1. Pathogenesis
Intradural spinal epidermoid tumors or cysts are rare, benign lesions that can arise when epidermal cells are abnormally displaced into the thecal sac. During embryogenesis, the central nervous system and skin both develop from ectoderm, which is why epidermal cells may grow within the thecal sac. These tumors can either be congenital or acquired. Congenital causes of spinal epidermoid tumors include: spina bifida, myelomeningocele, diastematomyelia, hemivertebrae, and syringomyelia. Acquired causes include: lumbar puncture, trauma, and prior surgery [3].
Lumbar puncture is a common, iatrogenic cause of epidermoid formation, but can theoretically be prevented with the use of styletted needles. However, in the pediatric population the stylet may be removed and therefore carries the risk of introducing epidermal cells in the dural sac [1]. Historically, the bore of the needle was greater than those in use today and therefore could displace a greater number of epidermal cells [29].
2. Clinical Presentation
Symptoms are partially dependent on the location of the tumor but can include: weakness, sensory disturbances, low back pain, painful radiculopathy, and bowel and bladder dysfunction. In the event of rupture of the epidermoid tumor’s capsule into the subarachnoid space, patients can present with acute, chemical meningitis [30]. A case series by Manno et al. [16] found that the majority of spinal epidermoid tumors formed in the lumbar spine (92%). However, they may form at other locations including the thoracic spine and cauda equina. Morita et al. [3] found that the mean time to presentation for acquired epidermoid tumors after the precipitating event was 9 years. Another study examined the growth rate of epidermoid tumors and found that they grow at a similar rate as normal skin cells do, which explains why there is a delayed presentation [31].
3. Imaging
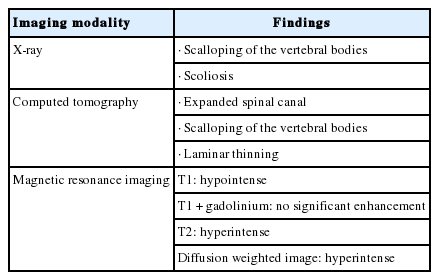
Table 2 summarizes the imaging findings of spinal epidermoids [17,29,32,33]. Since cerebrospinal fluid (CSF) appears hypointense on T1 and hyperintense on T2, multiple studies proposed using diffusion weighted imaging (DWI) as the most definitive MRI sequence to diagnose and monitor spinal epidermoid tumors [17,33]. On DWI, spinal epidermoid tumors appear hyperintense, which helps differentiate it from CSF, which is hypointense [17]. The differential diagnosis of these intradural, extramedullary tumors includes dermoid, meningioma, lipoma, and neurofibroma [29].
Radiographic imaging of spinal epidermoid tumors
4. Pathology
Pathological examination of epidermoid tumors reveals stratified squamous epithelium with an outer layer of collagenous tissue. The interior of the tumor is rich with cholesterol crystals, which form through desquamation of keratin from the epithelial lining. Epidermoid tumors can be differentiated from dermoid tumors by looking for adenexal structures, which are absent in the former and present in the letter (Fig. 3) [3,5].
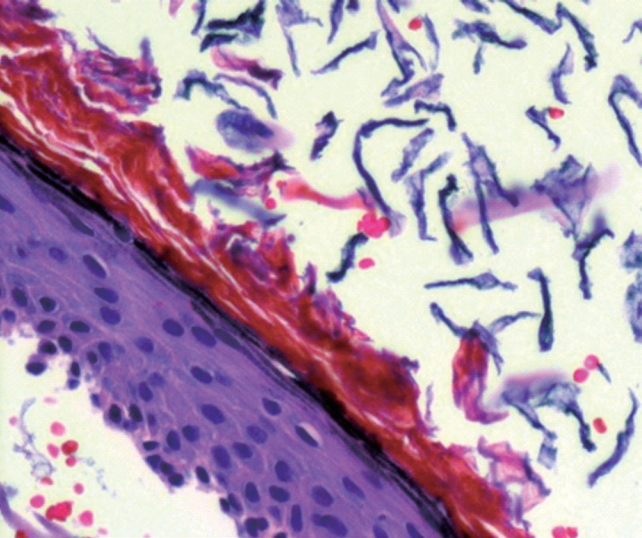
Histopathological examination of hematoxylin and eosin (H&E) stained sections shows the classic morphology of epidermoid cyst, including a cyst lining composed of keratinizing stratified squamous epithelium, and cyst contents comprising sheets of flaky keratin (anucleate squames). No dermal adnexal structures are present (H&E, × 200).
5. Treatment
In symptomatic patients, surgical resection is the primary treatment. Given that these tumors can be tightly attached to the surrounding neural tissue, particular care should be taken when dissecting to establish a plane with the nerve roots or spinal cord. When possible, total excision of the tumor should be performed, since recurrence or chemical meningitis may occur if residual tumor is left behind. However, if the capsule is adherent to the nerve roots, a STR may be appropriate to relieve mass effect. As expected, adhesions are more significant in redo surgeries for recurrent tumors. Patients should follow-up at regular intervals to monitor for recurrence even if total resection was performed [3]. In patients who have undergone multiple surgeries without complete resolution of symptoms or in those with extensive fibrosis, radiotherapy may be useful for palliation [5,34]. Intraoperative neurophysiological monitoring is often utilized when resecting spinal tumors. Given that epidermoid tumors may be tightly adherent to the surrounding neural tissue, EMG and somatosensory evoked potentials can be used to help guide the surgical resection of these tumors [15].
Spinal epidermoid tumors tend to present at an early age with a median age of diagnosis being 23. This is largely due to the fact that congenital deformities involving closure of the neural tube can lead to the aberrant displacement of epithelial cells within the dura. Furthermore, performing lumbar punctures without the use of a stylet in the pediatric population has further increased the incidence of these tumors in younger patients [1]. This highlights the importance of using correct technique when performing lumbar punctures as well as avoiding seeding of epithelial cells into the intradural space during spinal surgery. Nine cases were congenital (14%) and therefore epidermoid tumors should be included in the differential diagnosis for patients with spinal dysraphism who present with new onset lower back pain and leg pain with motor or sensory disturbances. The literature reviewed yielded a total of 26 epidermoids that were described as adherent. Of these, 20 patients underwent a GTR with 18 had positive outcomes after surgery. STR was performed 4 out of 26 adherent tumors with only 1 of these patients experiencing a positive outcome. This suggests that even in the case of adherent tumors, GTR provides better outcomes despite the expected risk of neurological deficits when dissecting away from the neural elements. However, there is no high-quality evidence of clinical studies that compare degree of resection for spinal epidermoids. Given the rarity of the tumor and the heterogeneity of presentation; literature reviews and case reports may guide practice in the immediate future.
CONCLUSION
Spinal epidermoid tumors are rare, benign tumors that are more common in women and younger patients. As seen in other intradural, extramedullary tumors, patients typically present with low back and leg pain, bladder and bowel dysfunction, muscle atrophy, and sensory disturbances. Many cases are acquired from trauma, surgery, or lumbar puncture (46%). MRI with DWI is particularly useful to confirm the diagnosis with the lesions appearing avidly hyperintense. The most common approach to resection involved laminectomies and intradural tumor resection. GTR provided better outcomes in the majority of cases even when the tumor was adherent to the surrounding spinal cord or nerve roots.
Notes
The authors have nothing to disclose.
SUPPLEMENTARY MATERIALS
Supplementary Table 1 can be found via (https://doi.org/10.14245/ns.1836014.007).