Arnold Chiari I Malformation with Tip-Toe Gait: A Case Report
Article information
Abstract
The Arnold-Chiari type I malformation has many symptoms such as headache, neck pain, gait impairment, abnormal movements or postures. But a few cases reported association of specific symptom including headache, neck pain, vertigo or ataxia. We report a case of 12 year-old boy presenting with tip-toe gait. Magnetic resonance imaging (MRI) study of brain and spine revealed underlying Arnold-Chiari type I malformation. This case shows that it is need for central nervous system evaluation in patients with changes of lower extremities tone.
INTRODUCTION
Arnold-Chiari malformation (ACM) is classified into types I and II according to cerebellar structure relative to the foramen magnum. Type I is defined as a decrease in the cerebellar tonsil, and type 2 involves the cerebellar vermis or medulla6,8).
After the first report by Chiari in 1891, many cases of ACM have been reported. The most common symptoms of this affliction are headache, neck pain, gait disturbance and abnormal movement or posture. Recurrent respiratory infection and abnormal development are also rarely reported. However, diagnosis of the disease without radiologic evaluations is difficult because there are no distinguishing symptoms of the disease1,6,11).
The authors observed a case of ACM I that did not exhibit the above symptoms except tip-toe gait; therefore, we report the clinical symptoms observed with electrodiagnostic studies, magnetic resonance images and genetic studies.
CASE REPORT
A 12-year-old male patient presented to our institute complaining of tip-toe gait for the previous ten years (Fig. 1). The patient was born via normal vaginal delivery with a weight of 3.2 kg and a gestational age of 36 weeks. The developmental course had been normal. The boy underwent surgery to correct a ventricular septal defect at four years of age, and his sister suffered from an atrial septal defect. His mother reported that the boy's maternal grandfather also had a tip-toe gait. The patient had not previously experienced abnormal motor or sensory deficits, pain or discomfort, nor did he have any problems walking or running. Neither he nor any of his family members have undergone ankle evaluation.
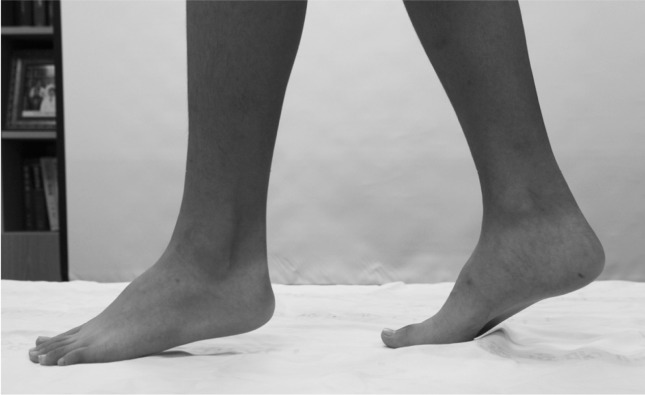
A clinical photograph of the patient shows tip-toe gait during standing and walking.
On physical examination, the motor and sensory abilities of both the upper and lower limbs were normal. No pathologic reflexes were observed, and the passive dorsiflexion and plantarflexion of both ankles were 0° and 50°, respectively. These results indicate reduced ranges of motion (ROM) of ankle dorsiflexion.
Other than those of the ankles, the ROM of all joints were within the normal limits. No muscular atrophy was observed in the upper or lower limbs, and no valgus or varus deformities were observed in either ankle. And neurologic examination about cerebellar function was performed and showed normal findings. The biceps, triceps and knee jerk responses were normal, exhibiting the double-positive reaction; however, both ankle jerks were slightly hypertonic as triple-positive reactions. The patient's heels did not contact the floor during walking.
At admission, the blood test and ankle radiologic X-ray results were normal, and the electrodiagnostic study including nerve conduction test, electromyography and somatosensory-evoked potential studies were all normal. Therefore we thought the cause of his tip-toe gait was less likely to peripheral nerve or orthopedic problems. So tests for determining upper motor neuron disease and magnetic resonance images (MRI) of the brain and whole spinal cord were conducted. The MRI of the whole spinal cord was normal; however, a brain MRI revealed that the cerebellar tonsil was about 7mm lower than normal. This finding suggested the diagnosis of ACM I (Fig. 2). To differentiate from hereditary spastic paraplegia (SPG), SPG3A and SPG4 gene studies were performed with normal results.

Midsagittal T1-weighted MRI of the brain shows herniation of the cerebellar tonsil 7 mm below the foramen magnum.
The only symptoms demonstrated by the patient were tip-toe gait, hyperreflexic ankle tone and limitation of ankle dorsiflexion. Surgery was therefore not recommended, and the patient was observed and followed-up with the Neuro-surgery Department. He also underwent applied physical therapy for his ankles, resulting in improvements.
DISCUSSION
The clinical symptoms of ACM I are headache, visual disturbance, tinnitus, dizziness, dysphagia or cerebellar ataxia; however, because these are not specific findings, a differential diagnosis from other diseases is difficult. Therefore, ACM I is typically discovered during evaluations of other brain and spinal cord diseases. Generally, diagnosis of the ACM I is difficult until aggravated symptoms appear, such as secondary dissociated anesthesia, scoliosis and limb weakness accompanying syringomyelia5,9).
The diagnostic criteria of ACM I differ according to the study. Because a greater than 3mm descent in the cerebellar tonsil below the foramen magnum is rare in normal adults, and a greater than 5mm descent in the tonsil below the foramen magnum induces symptoms, the criterion of Arnold-Chiari malformation is descent of the cerebellar tonsil more than 5 mm below the foramen magnum1,2).
The typical management of ACM is suboccipito-cervical decompression surgery. Indications of the need for surgery are severe headache, optic symptoms including diplopia, visual disturbance, paresthesia, ataxia, and dysphagia5,6,9,11). Surgery has a typical prognosis of greater than 80% improvement in ACM I11).
Unlike ACM II, with an average survival time of less than 2-3 years, there are instances of asymptomatic Type I ACM in adults. In these cases, surgery is not required as long as no symptoms appear. The patient in the reported case had only tip-toe gait and thus was not subjected to surgery. However, manifestation of abrupt neurologic abnormalities would indicate a need for surgery, so intermittent brain MRI and regular follow-up were prescribed.
Heel-to-toe gait is normally achieved in children around three years of age, after which tip-toe gait must be differentiated from habitual toe gait, congenital short tendo calcaneus, cerebral palsy, syringomyelia, spinal tumor, hematoma in a lower limb muscle, or neuromuscular diseases like muscular dystrophy, Charcot-Marie-Tooth disease and hereditary sensorimotor neuropathies7,10,12).
If the toe gait persists for 2-3 years, this gait pattern is not considered to be habitual. Therefore, the judgment of habitual toe gait should not be determined without evaluation for other diseases12). In addition, principles is ruling out clinical other disease. In the reported case, because the ACM I was diagnosed according to radiologic findings, a diagnosis of habitual toe gait was inappropriate10).
The authors suspected autosomal dominant hereditary SPG because of the maternal grandfather's distinguishing gait. The patient did not have ataxia, mental retardation or optic neuropathy, and the results of the electrodiagnostic and genetic studies were normal. Based on these findings, we ruled out hereditary SPG. Spinal cord disease was also eliminated using spinal MRI, ultrasonography ruled out an intramuscular mass, and electrodiagnostic studies eliminated neuromuscular or other neuropathic diseases.
Congenital short tendo calcaneus could not be completely ruled out. However, this affliction is only suspected if tip-toe gait is not improved during development. However, a brain MRI of our patient confirmed ACM I, thus eliminating the possibility of congenital short tendocalcaneus7).
The diagnosis of ACM I in the reported case was difficult because only tip-toe gait was observed with no other general symptoms of the disease. Children with tip-toe gait may also suffer from cerebral palsy, especially if the patient was born prematurely. Cerebral palsy cannot be completely ruled out even with a normal brain MRI. However, in children with cerebral palsy without delayed motor development, mild spastic quadriplegia in patients with tip-toe gait is observed due to an increased muscle stretch reflex in the knees and ankles. However, even in such cases, hypertonic ankle tone is very rare10). In comparison, pediatric ACM can result in non-specific symptoms, unlike that in adults.
Hypertone or spasticity is dysfunction within the central nervous system of descending pathway to and within the spinal cord causes a upper motor neuron syndrome. The spinal alpha motor neuron is considered to be final common pathway for expression of spasticity but the more complex motor pathways involved in the disorder the movements of spastic brain injury such as anoxic, toxic, or metabolic encephaolpathies and tumor, abscess, cyst, vascular malformations, infarction, hemorrhage or trauma of brain3). And Jose et al. reported that 10 patients showed spasticity as their important symptoms of Chiari malformation among 27 patients with Chiari malformation I4). Therefore we thought that in this case tip toe gait caused by ankle spasticity was related with ACM as brain lesion because many diagnostic examinations shows less possibility of peripheral neuropathy, genetic disease and orthopedic problems.
CONCLUSION
Pediatric patients with ACM can be shows atypical symptoms such as tip-toe gait and hypertonic ankles, therefore careful and active assessment is needed for correct diagnosis and proper treatment.