Recent Molecular and Genetic Findings in Intramedullary Spinal Cord Tumors
Article information

Abstract
The study of genetic alterations and molecular biology in central nervous system (CNS) tumors has improved the accuracy of estimations of patient prognosis and tumor categorization. Therefore, the updated 2021 World Health Organization (WHO) classification includes various diagnostic genes, molecules, and pathways for diagnosis, as well as histological findings. These findings are expected both to have diagnostic applications and to facilitate new targeted therapies that target tumor-specific genetic changes and molecular biology. Intramedullary spinal cord tumors (IMSCTs) are rare CNS tumors that are difficult to treat because they occur in eloquent areas. Although the genetic underpinnings of IMSCTs remain unclear compared to their intracranial counterparts, the genetic characteristics of these tumors are gradually being revealed. Here, we describe the major changes in the new 2021 WHO classification and review the major types of IMSCTs, with an emphasis on their clinical features and genetic alterations.
INTRODUCTION
Central nervous system (CNS) tumors, which arise from the brain and spinal cord, are classified into over 120 entities. World Health Organization (WHO) Classification of Tumors of the Central Nervous System has been revised on several occasions since its first publication in 1979 [1]. In the prior version of the WHO classification, CNS tumors were classified based on their histological findings with a diagnosis term and WHO grade (I–IV) embedded. However, recent comprehensive genomic studies revealed that an integrated diagnosis based on pathological and molecular findings more accurately predicted the prognosis than a diagnosis based on pathological findings alone. An integrated diagnosis with molecular information was first presented in the 2016 WHO classification [2,3], and the latest version was published in 2021 [4].
Although intramedullary spinal cord tumors (IMSCTs) are included in the category of CNS tumors, they are much less common than intracranial tumors and their molecular and genetic studies considerably lag behind those of intracranial tumors. IMSCTs account for 5%–10% of all spinal cord tumors, with ependymomas and astrocytomas comprising 80%–90% of IMSCTs [5-7]. Hemangioblastomas are the third most common IMSCTs, after ependymomas and astrocytomas [8]. Recent advances in molecular and genetic research have revealed that some infratentorial tumors, including IMSCTs, have different genetic characteristics from supratentorial tumors [9-11]. In this article, we discuss the major changes in the 2021 WHO classification of CNS tumors and present a narrative review of the literature on recent molecular and genetic analyses that characterize the major types of IMSCTs.
Informed consent was obtained from all subjects involved in the study. This study was approved by the Ethics Committee or Institutional Review Board of Nagoya University (2012-0067-19).
MAJOR CHANGES IN THE 2021 WHO CLASSIFICATION OF CENTRAL NERVOUS SYSTEM TUMORS
CNS tumors were conventionally classified based on histological findings. Molecular parameters were incorporated into the classification of CNS tumors (i.e., an integrated diagnosis) in the 2016 WHO classification of CNS tumors and its update in 2021 [2,3]. The process of an integrated diagnosis for CNS tumors is still developing in the 2021 WHO classification of CNS tumors, where various key diagnostic genes, molecules, pathways are applied to define entities, and the nomenclature was made more consistent and simpler by only including the location, age, or genetic modifiers of clinical utility. Modifier terms like “anaplastic” are not used because “grading within types” is applied. Thus, terms such as “diffuse astrocytoma, IDH-mutant,” “anaplastic astrocytoma, IDH-mutant,” and “glioblastoma, IDHmutant” in the 2016 WHO classification of CNS tumors are now simply classified as “astrocytoma, IDH-mutant grade 2, 3, and 4.” Adult-type diffuse gliomas, which were divided into 15 entities in 2016, have been classified into only 3 types in 2021. To standardize the 2021 WHO classification of CNS tumors with other non-CNS tumor classifications, the term “type” and “subtype” have been adopted instead of “entity” and “variant,” respectively. For example, meningioma is considered a single type in the 2021 WHO classification of CNS tumors with 15 subtypes.
The WHO CNS tumor classification has adopted an original grading system in line with the non-CNS tumor classification to facilitate grading across different entities [12]. In the prior WHO classification of CNS tumors, a CNS was given one diagnosis name and automatically assigned to one WHO grade. In the 2021 classification, CNS tumors are graded within types and the grading is written using Arabic numerals, not Roman. For example, astrocytoma is assigned to grade 2, 3, or 4 based on its histological and genetic findings in 2021. Nonetheless, the 2021 WHO classification has generally retained the ranges of grades used for tumor types in prior editions (grade I: curable if surgically removed, grade IV: highly malignant, leading to death without effective therapy). Notably, although grading was based on histological findings in the prior version of the WHO classification of CNS tumors, molecular findings are applied as biomarkers to assign grades within the tumor type in the 2021 WHO classification of CNS tumors. The WHO grading of CNS tumors is no longer a histological system. For example, CDKN2A/B in IDH-mutant astrocytoma and the TERT promoter mutation, EGFR amplification, and +7/-10 copy number changes in IDH-wildtype astrocytoma are enough information to assign grade 4 even if the tumor is histologically low-grade [13].
THE CLINICAL FEATURES AND GENETIC FINDINGS OF INTRAMEDULLARY SPINAL CORD TUMORS
1. Spinal Astrocytomas
Astrocytomas are the second most common IMSCTs observed in adults, but the most common in children [9]. Several recent studies have demonstrated a clear prognostic difference between high-grade (WHO grade 3, 4) and low-grade spinal astrocytomas (mainly pilocytic astrocytoma WHO grade 1 and diffuse astrocytoma WHO grade 2) [5-7]. Therefore, we discuss spinal high-grade and low-grade astrocytomas separately, with descriptions of the important genetic mutations in each category.
1) High-grade astrocytomas
Gross total resection (GTR) is almost impossible for most high-grade astrocytomas, and subtotal resection or biopsy followed by radiation and chemotherapy is performed as the standard treatment in practical settings. Even with such treatment, the prognosis of these tumors is unfavorable (Fig. 1). There is a survival advantage with an excision extent of 78% or higher in intracranial glioblastoma, comparable to total excision [14]. Even in spinal high-grade astrocytomas, the extent of excision might be correlated with the prognosis, although the exact threshold of the extent of excision that improves the prognosis has yet to be clarified [15,16]. The benefits of adjuvant radiation and chemotherapy are controversial, although they are often performed for patients with residual tumors [9,16-18].

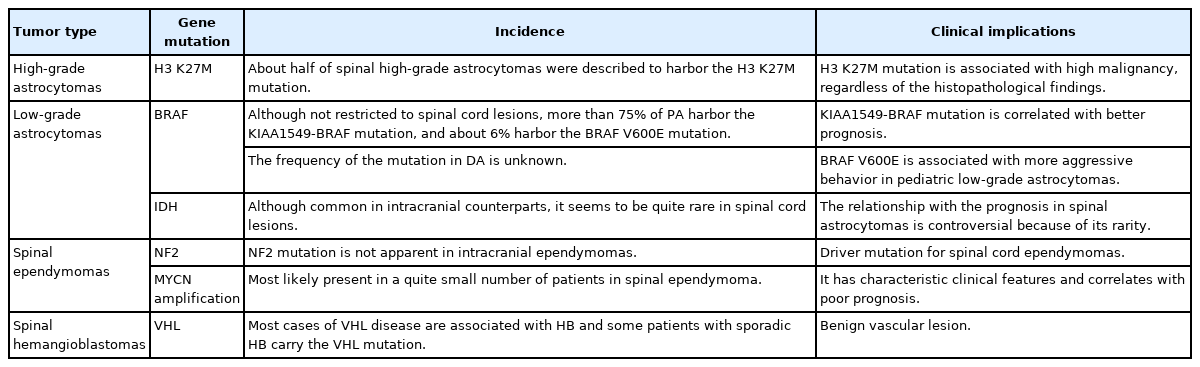
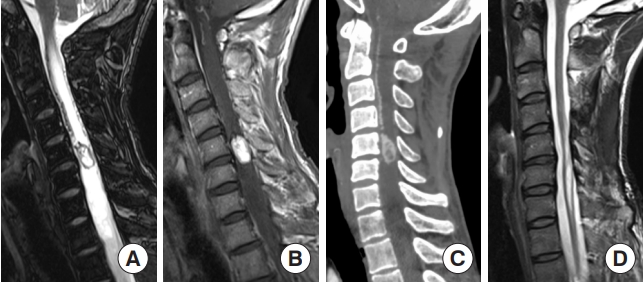
Diffuse midline glioma, H3K27-altered. (A) A 15-year-old male patient with neck pain and limb weakness for 2 months presented with an intramedullary tumor with ring enhancement on gadolinium-enhanced T1-weighted magnetic resonance imaging (MRI) from the medulla oblongata to the C4 level on MRI. (B) T2-weighted MRI showed extensive edematous changes. (C) The tumor was surgically removed, although MRI one month later showed residual tumor. A histopathological examination showed anaplastic astrocytoma; however, the H3 K27M mutation was found, and the final diagnosis was diffuse midline glioma, H3K27-altered. After surgery, the patient underwent radiotherapy and chemotherapy. (D) Fifteen months after the first surgery, MRI showed tumor regrowth, and the patient died 21 months after surgery.
(1) H3 K27M
The H3 K27M mutation of the H3F3A gene is the most important and well-established genetic mutation in high-grade astrocytomas (Table 1). Histones are major proteins that provide structural support for chromosomes. The histone tail (the N-terminal of the histone protein) plays an important role in the transcriptional regulation of DNA. Histone tail amino acids undergo various chemical modifications such as acetylation and methylation. The major histone is H3.1 in humans, and the histone subtypes referred to as histone variants, such as H3.2 and H3.3, are functionally encoded by a different gene. The K27M mutation changes lysine 27 to methionine in the N-terminus of the histone tail of the H3F3A gene, which mainly encodes the histone variant H3.3. This mutation is frequently detected in pediatric glioblastomas, diffuse intrinsic pontine gliomas (DIPGs), thalamic gliomas, and spinal cord astrocytomas [19-21]; however, it is not found in other CNS tumors or normal nerve tissues. This indicates that H3 K27M is a driver gene abnormality in brain stem gliomas and could be a powerful diagnostic marker of spinal diffuse astrocytomas [22].
Summary of intramedullary spinal cord tumors
In the 2016 WHO classification of CNS tumors, diffuse midline gliomas with the H3 K27M mutation were categorized as having a poor prognosis following the results of molecular diagnosis. Diffuse midline gliomas, characterized by a specific site mutation of H3 K27M, usually occur in the midline of the CNS, and this entity was renamed “diffuse midline glioma, H3 K27-altered” to reflect the fact that other changes (e.g., EZHIP protein overexpression) can define this entity in addition to the previously recognized H3 K27 mutations in the fifth edition of the WHO Brain Tumor Classification (2021) [4].
According to recent studies of comprehensive molecular profiling of CNS tumors, approximately 50%–60% of high-grade spinal astrocytoma cases have the H3F3A K27M (H3 K27M) mutation, as do DIPGs and thalamic gliomas [6,19,21-25].
H3 K27M-mutant spinal cord gliomas are highly malignant tumors according to the WHO classification; however, their clinical manifestations, imaging characteristics, chemotherapy, and appropriate surgical treatment have not yet been well-elucidated due to their rarity. At present, there are few reports about the diagnosis and treatment of H3 K27M-mutant spinal cord gliomas [24,26-28]. Although H3 K27M-mutant diffuse midline gliomas have currently been classified as WHO grade IV, recent studies on high-grade gliomas of the spinal cord have not revealed a clear prognostic difference between the prognosis of H3 K27 wild-type cases and H3 K27M mutant cases [6,24,28]. However, high enhancer of zeste homolog 2 (EZH2) expression and H3 K27me3 loss may be associated with a poor prognosis [22,29]. The interaction between the H3 K27M mutation and polycomb repressive complex 2 is promoted by EZH2. This interaction results in an overall reduction of H3 K27me3, as observed in tumors other than spinal cord gliomas. Ishi et al. [22] found that the combination of H3 K27me3 status and EZH2 expression had prognostic value for WHO grade 2–4 diffuse spinal cord gliomas. Maeda et al. [30] reported that mutant allele-specific imbalance was associated with significantly higher Ki-67 index and poorer survival, and related to downregulation of H3 K27me3 modification.
2) Low-grade astrocytomas
The majority of spinal cord low-grade astrocytomas are grade 1 pilocytic astrocytomas and grade 2 astrocytomas. These tumors have been reported to have a better prognosis than high-grade astrocytomas [6,31]. Surgical resection is the mainstay of treatment for patients with low-grade spinal cord astrocytoma with the intention of maximizing resection and avoiding long-term neurological dysfunction. GTR may be possible in cases with a clear tumor-parenchyma interface (Fig. 2). It is difficult to achieve GTR with more invasive tumors, such as grade 2 astrocytoma, although a better prognosis is expected than with high-grade astrocytoma because of their slower growth pattern (Fig. 3).
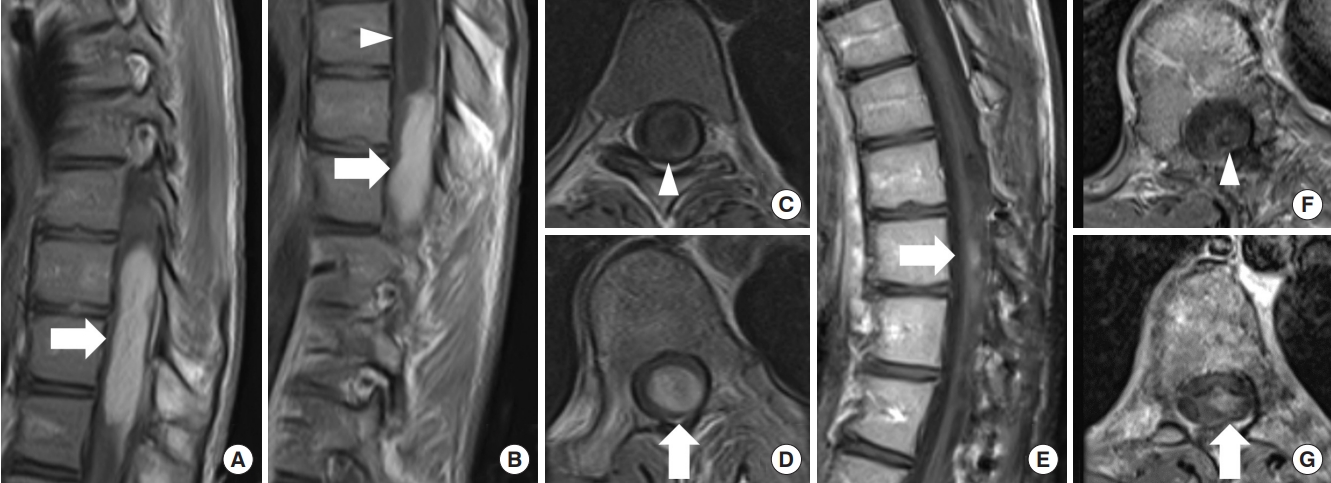
Pilocytic astrocytoma, World Health Organization grade 1. (A–D) An 18-year-old man scheduled for surgery for scoliosis revealed a mass lesion at T8–12 with syrinx. The tumor was shown in contrast by gadolinium-enhanced T1-weighted magnetic resonance imaging (MRI) (arrow: tumor, arrowhead: syrinx). During the waiting period for surgery, the patient suffered from sudden onset of paralysis of the lower limbs and urinary retention, and emergency surgery was performed. The tumor had a relatively distinct surgical plane that separated it from the surrounding spinal cord parenchyma, and almost the entire tumor could be removed. Thirty-one months later, MRI showed that a tiny contrast-enhancing lesion was still present (E, G) and the syrinx resolution (F). At 40 months postoperatively, the patient’s neurological symptoms were stable.
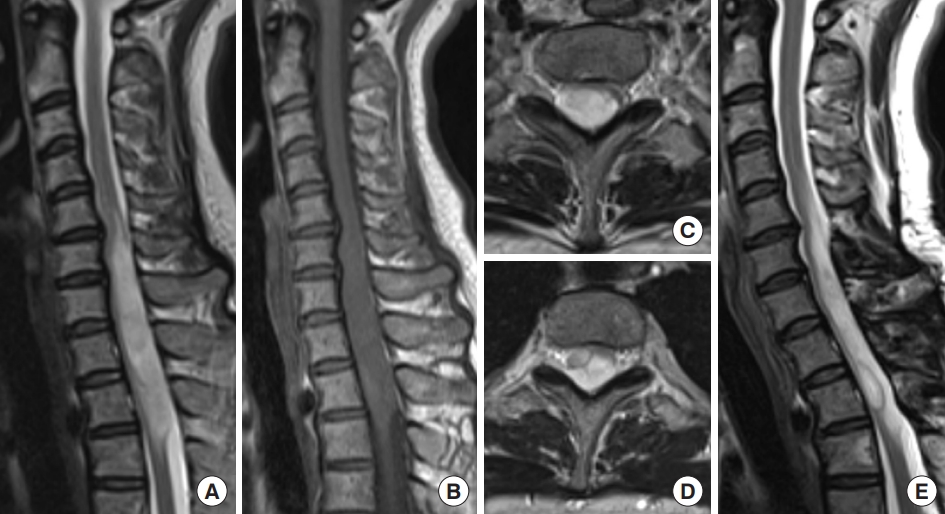
Diffuse astrocytoma, World Health Organization grade 2, IDH1 R132H mutant. A 42-year-old woman experienced numbness in both lower extremities for about 3 years and numbness in the right upper extremity and muscle weakness in the right lower extremity for 2 months. Magnetic resonance imaging (MRI) revealed an intramedullary spinal tumor at the C5–T3 levels. The tumor diffusely expanded with high signal intensity on T2-weighted MRI (A, C, D) and without any enhancements on gadolinium-enhanced T1-weighted MRI (B). It was difficult to identify a distinct surgical plane for the tumor, and partial removal was achieved. The tumor was diagnosed as diffuse astrocytoma, IDH1 R132H mutant. Forty-two months later, MRI showed no evidence of tumor growth (E), and her symptoms were stable.
(1) BRAF
Two major mutations have been noted in BRAF: a fusion oncogene between BRAF and KIAA1549 (KIAA1549-BRAF), and the substitution of valine to glutamate at position 600 (BRAF V600E) [32]. These genetic mutations have been reported to activate the mitogen-activated protein kinase (MAPK) pathway, which is associated with tumorigenesis [33,34]. Pilocytic astrocytomas harboring the BRAF V600E mutation account for about 5%–15% of all cases [35], and the KIAA1549-BRAF fusion gene is more likely to be detected, especially in infratentorial lesions [32,36,37] (Table 1). In a study including 10 cases of grade 1 spinal cord pilocytic astrocytomas, there were 3 cases with BRAF-KIAA1549 translocation and 5 cases with BRAF copy number gain [38]. Another study that included 26 grade 1 spinal cord pilocytic astrocytomas revealed that 10 patients harbored the KIAA1549-BRAF mutation and 1 patient harbored the BRAF V600E mutation [31].
BRAF mutations have also been reported in grade 2 spinal cord astrocytomas. In a study including 10 cases of grade 2 spinal cord diffuse astrocytomas, 1 case had BRAF-KIAA1549 translocation and 2 cases had BRAF amplification [38]. Another study that included 17 grade 1 spinal cord pilocytic astrocytomas revealed that 2 patients harbored the BRAF V600E mutation [31]. Low-grade gliomas with BRAF mutations can be classified into new tumor types according to the 2021 WHO classification. Reflecting the practical and conceptual importance of separating pediatric gliomas from other diffuse gliomas, 2 additional groups were added: pediatric diffuse low-grade gliomas and pediatric diffuse high-grade gliomas. “Diffuse low-grade glioma, MAPK pathway-altered” in the group of pediatric diffuse low-grade gliomas is defined as a pediatric glioma with broad histologic features, including astrocytic, oligodendroglial, or mixed morphology that shows activation of the MAPK pathway, such as BRAF mutations [4].
The influence of BRAF mutations on the prognosis remains controversial. Some studies have shown that the KIAA1549-BRAF fusion is associated with improved prognosis in pediatric low-grade astrocytomas [36,39,40], while another report revealed that the absence of the KIAA1549-BRAF fusion did not contribute significantly to the prognosis of spinal cord grade 1 pilocytic astrocytomas [31]. The BRAF V600E point mutation is thought to be associated with more aggressive behavior in pediatric low-grade astrocytomas [40].
(2) IDH
Isocitrate dehydrogenase (IDH) mutations were first identified in 2008 in intracranial glioblastoma [41] and in > 80% of WHO grade 2 and 3 cases [42,43]. Because IDH mutations are associated with the prognosis of intracranial gliomas [43,44], IDH mutations are considered clinically significant, and the 2021 WHO classification classifies the common diffuse gliomas of adults into 3 types: “astrocytoma, IDH-mutant”; “oligodendroglioma, IDH-mutant and 1p/19q-codeleted”; and “glioblastoma, IDH-wildtype.” [4] However, IDH mutations are extremely rare in spinal gliomas and their incidence in spinal cord gliomas is not well understood [32,45] (Table 1). According to the results of immunohistochemistry and Sanger sequencing for 120 midline gliomas, including 35 spinal gliomas, 61 patients tested positive for the H3 K27M mutation, while only 2 cases exhibited the IDH1 R132H mutation [46]. In another study examining the molecular characteristics of 83 spinal gliomas, there were no IDH1 mutations, although H3 K27M mutations were found in 35 cases [6]. Furthermore, the results of the genetic analysis of spinal cord gliomas by next-generation sequencing (NGS) have been recently reported. NGS for 61 intramedullary astrocytomas including 17 grade 2 diffuse astrocytomas, revealed 2 cases of IDH mutations [31]. In another study of NGS for 26 spinal astrocytomas, there were 2 IDH mutation cases [7]. Thus, spinal cord gliomas are less likely to harbor the IDH mutation than intracranial gliomas.
In addition, the majority of IDH gene mutations in intracranial gliomas are IDH1 R132H [43]; however, the IDH mutation variants found in spinal cord gliomas may be different from those found in intracranial gliomas. A recent retrospective study further supports this hypothesis. In this study of IDH1 R132H mutant gliomas and noncanonical IDH-mutant (not IDH1 R132H) gliomas, none of the 166 IDH1 R132H mutant gliomas included an infratentorial region, while nine of 155 (5.5%) noncanonical IDH-mutant gliomas were infratentorial regions [47]. Intracranial gliomas and infratentorial gliomas including the spinal cord seem to have different genetic underpinnings. Even though IDH-mutant spinal gliomas are very rare, several spinal gliomas with variants other than IDH1 R132H have been reported, and IDH1 R132H mutations are probably not so prevalent in spinal gliomas. Konovalov et al. [48] reported 5 cases of spinal cord astrocytomas with IDH mutations: 2 had IDH1 R132H mutations, while 1 had an IDH1 R132G mutation. In addition, the remaining 2 cases had translocations at positions 82 (Arg → Lys, R82K) and 76 (Ile → Thr, I76T) of the IDH1 gene, which had never been described in intracranial gliomas in the past. In another study, Takai et al. [49] reported one case of spinal astrocytoma with an IDH1 R132S mutation, and we previously reported 2 cases with IDH1 R132C and IDH1 R132H mutations, respectively [45]. There is also a report of spinal cord glioma with IDH2 R172 [31]. Even though IDH-mutant spinal gliomas are very rare, several spinal gliomas with variants other than IDH1 R132H have been reported, and IDH1 R132H mutations are probably not very prevalent in spinal gliomas. Therefore, Immunohistochemistry using an anti-IDH1 R132H antibody, which is commonly used for intracranial gliomas, is not sufficient for spinal gliomas, and genetic testing is desirable.
The prognostic impact of IDH mutations in spinal cord gliomas is controversial [7,31,45,49,50]. Because IDH mutations are rare in spinal gliomas, the current knowledge of IDH mutations and the associated prognosis is inadequate. However, the prognosis of IDH-mutant spinal gliomas does not seem to be different depending on the IDH variant. A study of IDH variants and patients’ prognosis found no significant difference in prognosis between IDH1 R132H mutant glioma and noncanonical IDH-mutant glioma [47]. The clinical significance of IDH mutations in spine gliomas needs to be confirmed in a larger cohort.
2. Spinal Ependymomas
Spinal ependymomas are the most common IMSCTs [9,51,52]. Almost all ependymomas are benign tumors with clear tumor borders, and long-term survival can be expected by targeting GTR (Fig. 4) [8,53]. Therefore, the long-term functional prognosis should also be considered in the treatment [54-57].
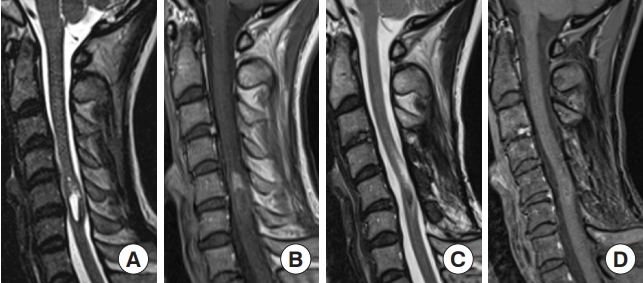
Spinal ependymoma. A 44-year-old woman who had been treated for multiple intracranial meningiomas developed numbness in her right upper extremity. Magnetic resonance imaging (MRI) revealed an intramedullary tumor at the C5–6 level with an enlarged spinal cord. (A) T2-weighted MRI showed a hyperintense tumor with a syrinx and a partially hypointense lesion reflecting hemosiderin. (B) The tumor was shown in contrast by gadolinium-enhanced T1-weighted MRI. Tumor removal was performed via a posterior approach, and gross tumor resection was achieved because the surgical plane of the tumor was clear. (C, D) T2-weighted MRI 67 months after surgical removal showed low intensity that reflected hemosiderin, whereas there was no evident tumor recurrence and no enhancement on gadolinium-enhanced T1-weighted MRI.
1) NF2
Neurofibromin (NF2) gene mutations are driver mutations for spinal cord ependymomas and appear to be the most prevalent genetic mutations in spinal ependymomas [58] (Table 1). Pajtler et al. [10] analyzed about 500 ependymomas, including 47 spinal lesions and revealed that most spinal ependymomas had a loss of the 22q locus, which harbors the NF2 gene, although NF2 mutations were not seen in intracranial ependymomas. In another study, 47% (9 of 19) of spinal ependymomas had NF2 mutations [9,59]. NF2 is a tumor suppressor gene, and aberrations of the NF2 gene make cells less responsive to contact inhibition, thereby promoting tumorigenesis [58].
2) MYCN amplification
“Spinal ependymoma, MYCN-amplified” is a new category in the 2021 WHO classification [4]. Although MYCN-amplified ependymomas are very rare, they are associated with aggressive behavior and unfavorable outcomes [60,61]1 (Table 1). Ghasemi et al. [62] investigated 13 MYCN-amplified ependymomas, of which 10 were WHO grade 3 and 3 were WHO grade 2 on histopathological examination. Compared to other subtypes of ependymomas, these groups had worse median progression-free survival (17 months) and median overall survival (87 months). These tumors were also characterized by a favored location of the cervical and thoracic spine, and were predominantly intradural and extramedullary [63]. The presence of diffuse leptomeningeal spread and dissemination has also been revealed as a distinctive feature of these tumors. Further study is required to develop new strategies to improve the prognosis of patients with MYCN-amplified spinal ependymoma.
3. Spinal Hemangioblastomas
Spinal cord hemangioblastomas are benign vascular lesions and constitute the third most common IMSCTs [8]. Surgical resection is recommended in cases of symptomatic lesions or lesions that appear to be growing on repeat imaging studies. Because hemangioblastoma generally shows a well-defined tumor border that allows GTR, radiotherapy has a limited role [8,64]. Although these are vascular-rich tumors, intraoperative hemorrhage is generally not a problem due to the availability of techniques such as temporary intraoperative arterial occlusion and preoperative embolization (Fig. 5) [8,65]. Although hemangioblastoma regrowth is very rare once the tumor is completely removed, patients with von Hippel-Lindau (VHL) disease may show multiple hemangioblastomas with new lesions repeatedly arising.
Hemangioblastoma. A 44-year-old man presented with numbness in the right upper extremity and right paralysis. A radiological examination revealed a tumor at the C5 level. (A) T2-weighted MRI showed a well-defined tumor with a syrinx and flow void. (B) The tumor was strongly shown in contrast with gadolinium-enhanced T1-weighted magnetic resonance imaging (MRI). (C) Dynamic computed tomography angiography also showed a strongly contrasted tumor with dilated feeder and drainer. Preoperative angiography and feeder occlusion were performed, and the tumor was totally resected microscopically via a posterior approach. Immediately after surgery, the patient’s symptoms improved. (D) Thirty-six months later, MRI showed no recurrence of the tumor and the syrinx had collapsed.
1) VHL
Approximately 20% to 40% of patients who develop hemangioblastomas have VHL disease [66], which is an inherited disorder that causes multiple tumors and cysts in various parts of the body (Table 1). Glasker et al. [67] reported that 94% of VHL disease-associated hemangioblastomas harbor VHL mutations and 62% exhibit loss of heterozygosity (LOH) at the VHL locus (3p25-56). By contrast, of 13 sporadic hemangioblastomas, 23% expressed germline mutations in VHL and 50% had LOH of the VHL locus [67]. The VHL gene encodes an E3 ubiquitin ligase that targets hypoxia-inducible factor-1a (HIF-α), which is known to be a regulator of vascular growth [9]. Mutations or deletions of the VHL gene cause cells to be unable to adequately degrade HIF-α, leading to vascular proliferation. Activated HIF-α and vascular endothelial growth factor were found to be correlated and increased in VHL mutant cells [32,68].
CONCLUSION
The discovery of the genetic and molecular mechanisms of CNS tumors is beginning to impact the management of intracranial tumors, with improved predictions of prognosis and availability of targeted therapy. The 2021 WHO classification has been modified to reflect these facts. However, the genetic underpinnings of spinal cord tumors remain less well understood as those of their intracranial counterparts due to their rarity and difficulty in treatment because of their location in eloquent areas. Molecular and genetic differences exist between tumors located in the spinal cord and intracranial regions, even within the same pathological type. Therefore, further genetic studies on IMSCTs are warranted in order to develop novel therapies and improve the prognosis of patients suffering from these challenging tumors.
Notes
Conflict of Interest
The authors have nothing to disclose.
Funding/Support
This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Author Contribution
Conceptualization: YN, YN, FO, MT; Data curation: YN, KE, JY; Project administration: YN, MT; Writing - original draft: YN, KE, JY; Writing - review & editing: YN, YN, SH, FO, MT, RS.