INTRODUCTION
Spinal cord injury (SCI) is the traumatic or nontraumatic damage to the spinal cord, causing impairment of motor function and leading to morbidity or permanent paralysis [1]. In today’s world, SCI is still a significant source of illness and mortality. Acute traumatic SCI affects people worldwide, with an annual incidence of 15–40 cases per million [2]. SCI is particularly concerning on a social level because it primarily affects young, otherwise healthy people, with injuries happening most frequently in those between the ages of 15 and 25 [3]. Creating effective recovery treatments requires a thorough understanding of SCI pathophysiology, stages, and diverse wound recovery mechanisms [4]. The major reason of SCI progression is the death of these different types cells relevant to spinal cord structural and functional homeostasis, initiated by numerous crosstalk between different cell death pathways. Many cell types interact in normal spinal cord physiology, including astrocytes, neurons, microglia, and oligodendrocytes. These multicellular interactions are disrupted and disorganized after an SCI, resulting in slowed spinal healing [5]. The critical process of neuroinflammation, which is connected to SCI, is implicated in neutrophils, microglia, macrophages, astrocytes, dendritic cells (DCs), B- and T-lymphocytes, as well as molecules including cytokines and prostanoids [6]. It involves the death of spinal cord neurons and associating glial cells like astrocytes, oligodendrocytes, and microglias [7]. Neuronal and glial cell death ultimately catalyzes axonal degeneration, accelerating SCI progression. Apoptosis, autophagy, and necrosis are well established in SCI. They are welldocumented in helping axonal degeneration also [8]. New cell death pathways include common ones like ferroptosis, mitoptosis, parthanatos, and pyroptosis, and rare ones like oxieptosis, alkaliptosis, and autoschizis, are being discovered in the pathogenesis and progression of SCI [9].
Due to their activation, SCI inevitably results in functional decline. The severity of subsequent damage caused by a cascade of cellular and molecular processes initiated by the primary trauma determines the fate of SCI [10]. After a human SCI, the necrotic and apoptotic cell death processes are known to occur. The contribution of autophagy in SCI is also well established [11]. The spinal cord's diameter is relatively small, and even a tiny transverse expansion of initial damage would result in a more significant disconnection between the brain and the spinal cord below the lesion site. Therefore, developing strategies to limit the secondary degenerative processes would be of utmost importance in SCI research. This review aims to summarize all programmed and non-programmed cell death (non-PCD) pathways along with their molecular mechanisms, crosstalks and Involvement in the progression of the pathophysiology of SCI to pave the way for developing effective treatment strategies.
CELL DEATH PATHWAYS AND THEIR RELEVANCE IN THE PROGRESSION OF SCI
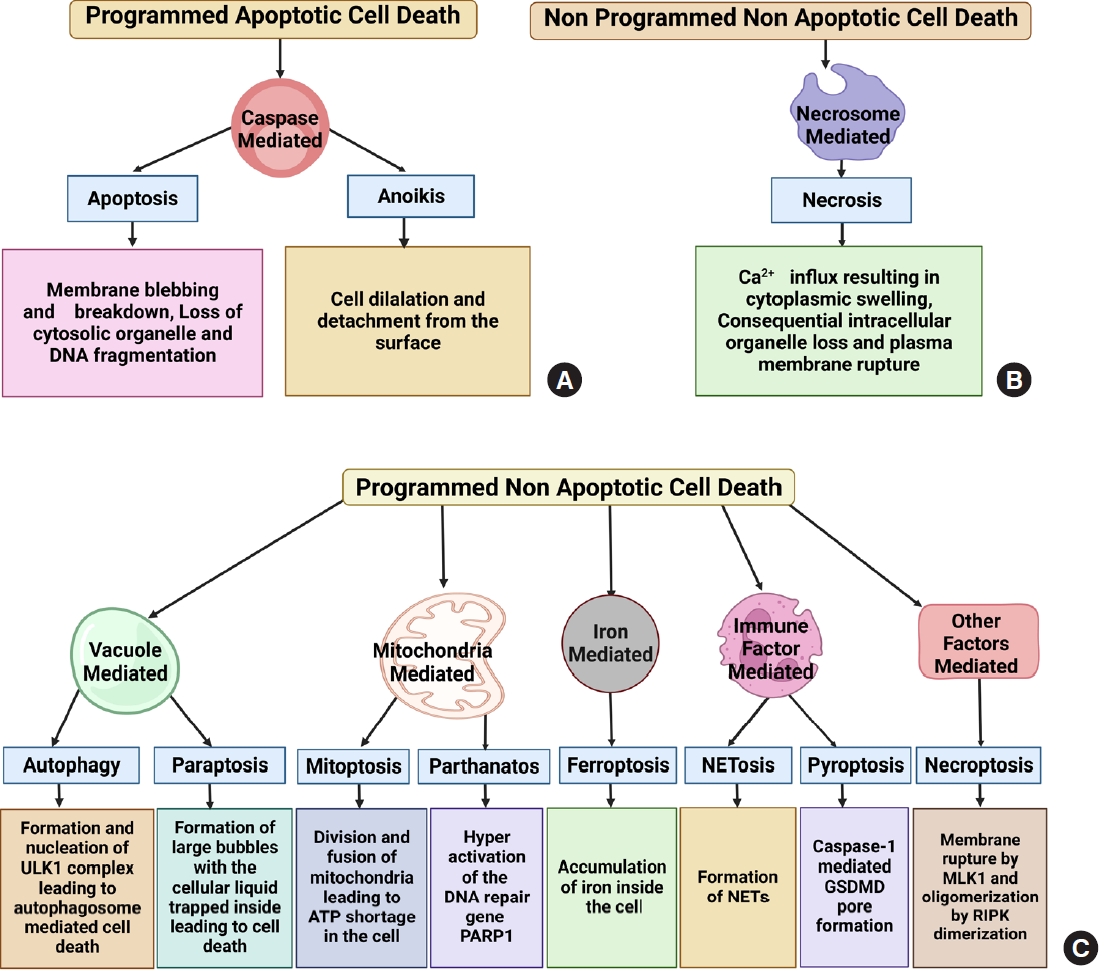
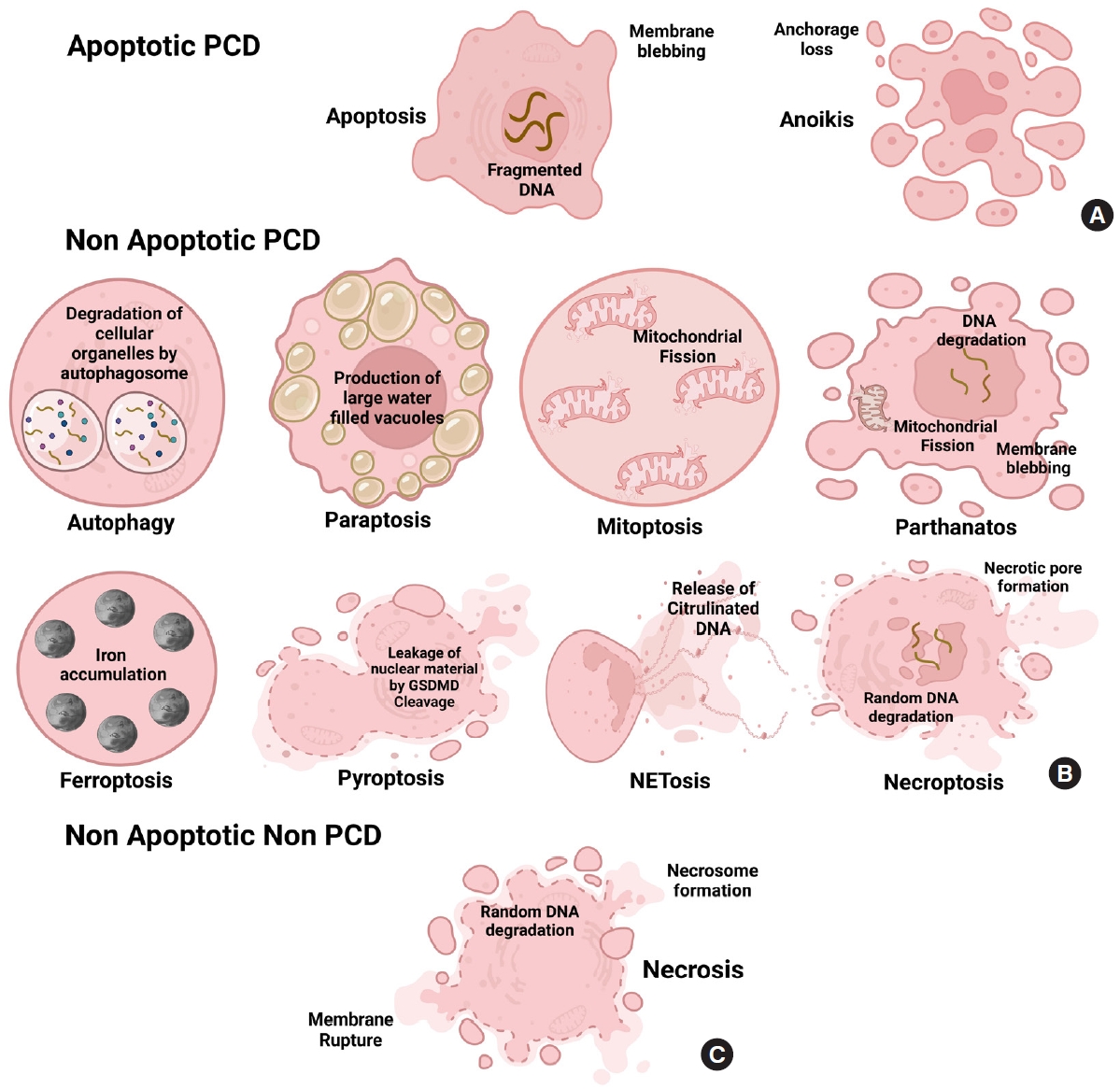
Cell death is the biochemical process by which a cell loses its ability to maintain its cellular morphology and homeostasis and serve its functions. It can be programmed when a cell becomes too old to continue serving its normal function or can be nonprogrammed, spontaneously induced by specific mechanical, ischemic or chemical trauma, causing the cell to die spontaneously. On one side, cell death is essential for a cell to cease its morphological and functional features through complicated programmed or nonprogrammed interactions, leading to organismic homeostasis. On the other hand, it can be detrimental as it can promote inflammation at the death site and affect other healthy cells to die, making it a double-edged sword [12]. The hallmarks of multiple cell death modes are identified and fitted into a basic classification framework, where cell death entities are classed as programmed or non-PCD, depending on their signal dependency. PCD is categorized into 2 types: apoptotic and nonapoptotic cell death. Programmed apoptotic cell death causes apoptosis and anoikis showing membrane blebbing, mitochondrial disruption, and cell detachment from the surface. Programmed nonapoptotic cell death includes vacuolepresenting cell death (autophagy, entosis, methuosis, and paraptosis), mitochondria-dependent cell death (mitoptosis and parthanatos), iron-dependent cell death (ferroptosis), and immunomodulatory cell death (pyroptosis and NETosis). Non-PCD includes necrosis which is vital in injury progression. Cell death is vital in disease development, notably cancer and injuries [13,14]. All cell death pathways show a plethora of morphological changes like the formation of fluid-filled bubbles inside the cell (paraptosis), mitochondrial disruption (mitoptosis and parthanatos), accumulation of iron (ferroptosis), formation of extracellular traps and gasdermin D mediated cleavage formation (NETosis and pyroptosis) or spontaneously induced (necrosis). Understanding cell death is vital for understanding how some illnesses progress, which leads to new treatment development (Figs. 1, 2).
Cell death pathways play a major role in SCI pathogenesis and progression. Initial tissue injury results in secondary injury, which further damages the spinal tissues chemically and mechanically, causes neuronal excitotoxicity because the calcium level in the cells is too high, and increases reactive oxygen and glutamate levels [15]. These events result in brain/spinal cord dysfunction by harming the underlying proteins, phospholipids, and nucleic acids [16]. The secondary injury phase, which follows the main damage phase and lasts several weeks, reflects multifeatured pathological processes. Caspase-mediated cell signaling, ischemia, vasculopathy, hydrops, excitotoxicity, ionic imbalance, inflammation, lipid peroxidation, free radical generation, demyelination, Wallerian degeneration, microglial scarring, and cyst formation are all clinical manifestations of secondary injury [16]. Ischemia is one of the major factors in disease progression, which can develop very once after traumatic SCI, and if it goes untreated, further damage may start within the first 3 hours and last for at least 24 hours [17]. In addition, monocytes, neutrophils, T and B lymphocyte cells, and macrophage infiltration occur due to blood vessel breakage, which causes bleeding in the spinal tissues. This phenomenon is also associated with the release of inflammatory cytokines such as interleukin (IL)-1α, IL-1β, IL-6, and tumor necrosis factor (TNF) after 6 to 12 hours following damage [18]. Inflammation of neurons is promoted by immune cell penetration and inflammatory cytokines [18]. Secondary injuries come in 3 forms: acute, subacute, and chronic, followed by primary injury [19].
Further, multiple inflammatory mediators, such as leukotrienes, bradykinin, prostaglandins, platelet-activating factors, and serotonin, are present in higher concentrations in the injured area [20]. Cell death is the cell’s final event, which can be segregated into 2 forms; apoptosis and necrosis. Apoptosis is the PCD pathway; nowadays, many PCDs are discovered [21]. PCD is crucial for getting rid of unwanted and damaged cells and can serve as a defense; it is also linked with CNS disorders and SCI [22]. On the other hand, apoptosis is a known physiological process that usually occurs and may be crucial in secondary SCI. The secondary injury after SCI is thought to be caused by the continuation of cellular destruction through apoptosis, and the longterm neurological deficits after SCI may result from a wide range of apoptosis of neurons and oligodendrocytes in the injured spinal cord [23].
DIFFERENT CELL DEATH PATHWAYS ASSOCIATED WITH SCI
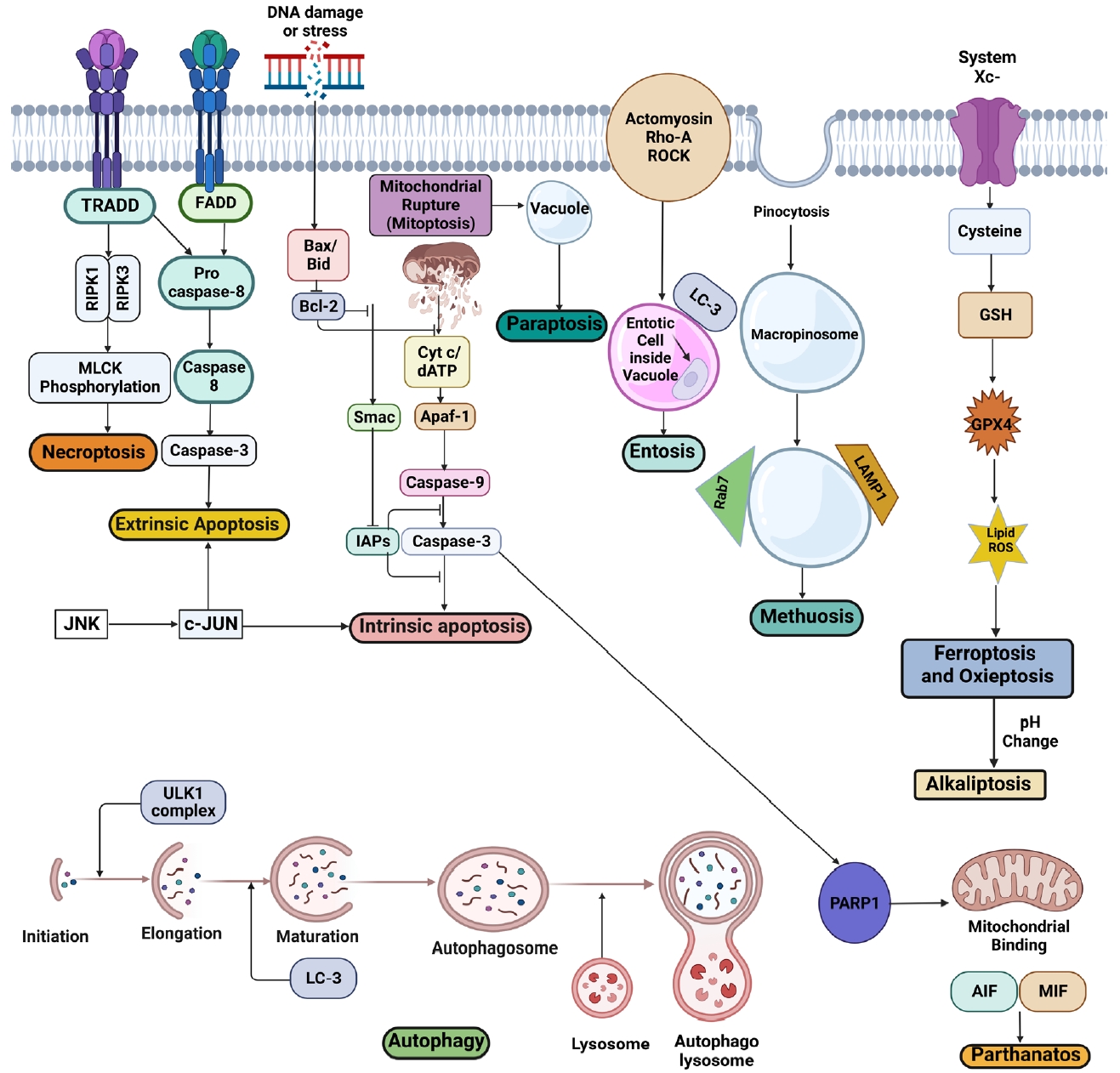
SCI pathophysiology is characterized by blood-spinal cord barrier collapse and breakdown, transmigration of immune cells, rupture of cellular axons and membranes, and myelin disintegration [6]. Some key signaling pathways, including CDK1/E2F1, AMPK/SIRT1, JNK/c-JUN, and Wnt-β catenin signaling pathways, are already involved in regulating apoptotic activity in SCI [24]. Recently, programmed and non-PCD has been recognized as a significant process after SCI. Several kinds of cell death pathways, including apoptosis, autophagy, ferroptosis, paraptosis, netosis, pyroptosis, and necroptosis, have been found in the direct link in the progression of primary and secondary SCI [9]. Here we will discuss them in more depth and detail (Fig. 3).
1. Programmed Apoptotic Cell Death in SCI
1) Apoptosis in SCI
Apoptosis is the most typical form of PCD, where cells die systematically in a signal-mediated manner, influenced by a cysteine protease caspase family-mediated cell death leading to planned self-destruction of the cell [25]. It is highly relevant to the progression of SCI. It is immunologically silent that occurs either by intrinsic (mitochondrial), extrinsic (death receptor [DR] triggered), or granzyme/perforin pathway-mediated mechanisms [26]. Inflammatory mediators, free radicals, and excitotoxins are the chemicals that induce necrosis or apoptosis [23]. They feature positional organelle loss, membrane blebbing, cell shrinkage, and DNA fragmentation after condensation [27]. A wide variety of apoptosis in oligodendrocytes and neurons in the damaged spinal cord may produce long-term neurological abnormalities after SCI. Therefore better knowledge is needed to furnish innovative treatment strategies [28].
In recent years, more studies have been done on the role of apoptosis in SCI. Apoptosis is seen both in injured human spinal cords and in animal models. The rat spinal cord showed apoptosis in astrocytes, neurons, microglia, and oligodendroglia [29]. Apoptotic oligodendrocytes are seen in white matter longitudinal filaments [30]. Apoptosis-induced damage to type 3 collagen of endoneurium leads to Wallerian degeneration of the neurons of the spinal cord leading to impairment of neurotransmission and motor functions [31]. Neuronal apoptosis was identified 4 hours postinjury and peaked 8 hours later; in glial cells, it peaked 24 hours later; in oligodendrocytes, in the white matter, it peaked 8 days later. Microglial apoptosis was the least frequent at 24 hours and 5 days after injury but rose quickly and peaked at 8 days [32]. The bulk of apoptotic cells may cluster near the damaged spinal cord’s center, explaining why the lesion area continuously enlarges [23]. Apoptosis is detected in oligodendrocytes with Wallerian degeneration in the chronic phase of SCI [33]. It involves activating components of both downstream and upstream origin in the executioner caspase-3 mediated apoptotic pathway after SCI in rats [34]. Following SCI in animals, the Fas DR pathway was revealed to be significant in microglial, oligodendrocydal, and neuronal apoptosis [35].
Apoptosis is induced by Fas-mediated cysteine protease activation, leading to DNA proteolysis and damage by effector caspases [36]. The function of long noncoding RNAs (lnc-RNAs) and microRNAs (miRNAs) in SCI pathogenesis, including cell death, is also being investigated. MiR-137 targets mitogen-activated protein kinase 2 to inhibit apoptosis after SCI [37]. Using proteomics, Liu et al. [38] discovered Erp29. This critical protein may influence several genes involved in cell death and survival, including Erk and caspase, and ameliorate locomotor activity and function in the rat spinal cord transection model. Gu et al. [39] observed that cutting down long coding XIST RNA reduced neuronal death after SCI by regulating the PTEN/AKT/mTOR pathway and competitively binding miR-494. Another study identified the AKT/mTOR/PTEN signaling pathway implicated in neuronal death after SCI, perhaps through activating the mitochondrial system [40]. It was recently discovered that caspase recruitment domain family member 6 inhibits Caspase-3 signaling and may reduce apoptosis [41]. Understanding apoptosis’s cellular and molecular mechanisms may help identify specific therapeutic targets. Minocycline, CD95 (Fas) ligand antibody blockage, and glycol sphingolipid-induced inducible nitric oxide synthase blocking have all been found to reduce neuronal death and boost the effectiveness of cell transplantation techniques [42]. Zheng et al. [43] observed that miR-142-3p vanquishes apoptosis in rat SCI.
Progranulin deficiency promotes cellular death and neuroinflammation, compromising SCI healing [44]. Zhang et al. [45] discovered that elevated p38 was related to apoptosis and inflammation in a rat SCI contusion model. He also hypothesized that reducing apoptosis and inflammation with the p38 inhibitor SB203580 might help secondary SCI. According to previous research, apoptosis causes tissue lysis and damage post-SCI.
A recent study found that metformin increased β-catenin and brain-derived neurotrophic factor expression, reduced neuron loss and inflammation, and improved functional and motor recovery in rats with SCI [46]. Finding a way to conquer SCI-induced apoptosis has huge therapeutic ramifications. However, the specific pathways triggering apoptotic death of astrocytes, neurons, microglia, and oligodendroglia following SCI are yet unknown [47].
2) Anoikis in SCI
Anoikis is a form of apoptotic PCD; it occurs because of the detachment of cells from the extracellular matrix (ECM) and is extensively observed in the degradation of oligodendrocytes and Schwann cells [48].
Inappropriate or inadequate interactions between cells and the matrix trigger it. The altered cytoskeletal dynamics are believed to interact with a critical prosurvival effector integrin. It leads to impaired ECM remodeling and myelin degeneration which worsens the injury and delays recovery [49]. Cell anoikis impedes recovery by aggravating CNS damage and impairing synaptic plasticity and other CNS activities [50]. Immunosuppression and neuroinflammation are 2 significant factors that contribute to anoikis promoting injury progression. To fasten spinal cord regeneration, adult stem cells are generally injected into a damaged location with high inflammation and poor vascularization [51,52].
Moreover, the lack/absence of ECM leads to post-traumatic cavity formation. Several studies indicate that after transplantation, the number of surviving stem cells decreases considerably in SCI models because of the death of cells by anoikis [53]. In the injury, enzymes are released, which lead to dysfunction and detachment of the ECM cells leading to anoikis; SCI may trigger the neuronal damage by anoikis and enhance the SCI progression. Anoikis inhibition might be the effective strategy to protect the PCD-induced neuronal damages [50]. Biomaterials are already in the research pipeline to prevent this ECM anoikis in SCI. Laminins also help to prevent anoikis in SCI [54].
2. Programmed Nonapoptotic Cell Death in SCI
1) Vacuole-mediated
(1) Autophagy in SCI
Autophagy is a controlled process that is initiated to remove cellular proteins and organelles in significant quantities by transporting them into membrane-bound vesicles to be uptaken by lysosomes to form autophagolysosome to initiate autophagic breakdown. They are formed when vesicles merge with lysosomes, and their contents are destroyed by lysosomal enzymes [55]. Autophagy has been shown to accelerate cellular mortality by activating caspase-reliant apoptosis in specific cells [56]. The PI3K/Akt/mTOR signaling pathway is essential in autophagy [57]. Recent research suggests that the lysosomal compartment plays a protective function in the oxidative stress response [58]. Increased autophagy has been documented after SCI, and emerging evidence suggests autophagy may help preserve neuronal and astrocytic cells from death after SCI, supported by the presence of autophagosomes in cultured and wild-type neurons [59]. However, the influence of autophagy in post-SCI neurodegeneration has been hotly contested, with mixed results from earlier SCI investigations.
Autophagy has been proposed as a potential SCI treatment target by specific studies. Metformin has been shown to protect against SCI by increasing autophagy [60]. He at al. [61] cultured CNS neurons and increased autophagy that stabilized microtubules by degradation of SCG10 (superior cervical ganglion protein 10) and increasing axon development. Axon retraction was reduced, axon regeneration increased, and functional recovery improved in SCI. Astrocyte autophagy flux may increase neurological repercussions, neuronal death, and survival. Many researchers have attempted to relate autophagy with apoptosis. Inducing autophagy protects against apoptosis in mice with acute SCI [62]. Autophagy also protects neurons from endoplasmic reticulum (ER) stress. Therefore its breakdown during SCI may cause ER stress-induced neuronal death [63]. Autophagy may protect spinal cord neurons against apoptosis, which may help SCI neuron survival by incorporating beclin-1 [64]. A contrasting research report found that lowering autophagosome biogenesis enhances spontaneous functional recovery by reducing distant axonal degeneration in SCI patients [65].
Recent research found that LC3+ cells increased considerably at the scar site after the rat spinal cord hemisection model, showing that autophagic cell death often occurs in injured neuronal tissue after SCI [66]. As Purkinje cells die off, phosphorylated MAP1B accumulates in their axonal dystrophic swellings and binds to LC3 at high levels. Therefore, MAP1B-LC3 interaction may contribute to controlling LC3-associated autophagosomes in neurons, especially in axons, under physiological and pathological situations [67]. ABT888, a poly (ADP-ribose) polymerase inhibitor, has recently been shown to protect against SCI by suppressing autophagy [68]. Because of this, autophagy may have both protective and harmful features in SCI. Understanding autophagy’s function in SCI may lead to creating a pleiotropic therapy that targets several pathways and types during the degenerative phase of SCI [69].
(2) Paraptosis in SCI
Paraptosis is another form of PCD, where the cell swells and develops large bubbles or vehicles with the cellular liquid trapped inside and eventually dying off. It occurs because of an imbalance in redox or ion homeostasis [70]. Due to cytoplasmic vacuolation, a novel nonapoptotic and caspase-independent PCD is characterized by ER and mitochondrial dilatation [71]. Previous studies demonstrated a paraptotic response targeting the nucleus in response to paraptosis [72,73]. Some study suggests that cycloheximide and aryl hydrocarbon receptor-interacting protein-1 (AIP-1) may alter paraptosis [74]. Nutlin-3/bortezomib may also disrupt proteostasis to induce long-term structural/functional changes in the mitochondrial and ER, causing mitochondrial/ER stress and, ultimately, cell death via paraptosis [75].
As a consequence, nothing is known about paraptosis in SCI. Increased p44 expression in the CNS is connected to neuronal death and insulin-like growth factor 1 receptor activation through autophagy and paraptosis [76]. Active microglia may lead to neuronal death with characteristics like vacuolation after the caspase cascade has been halted [77]. Studying the activated microglial role in paraptosis might be an exciting new technique. While paraptosis research has advanced, many questions remain. Paraptosis has not been widely investigated in connection to SCI, indicating that additional study is required.
(3) Other minor vacuole-mediated cell death pathways in SCI
Two other critical vacuole-mediated pathways apart from autophagy and paraptosis may have some link in SCI progression. They are entosis and methuosis. Entosis is a novel and interesting nonapoptotic PCD where rather than a cell being swallowed after it is dead, one viable cell actively invades or is pushed into a neighboring cell and dies afterwards, making this process unique. The internalized cell resides in some vacuole structure. Entosis is not restricted to interactions between just 2 cells but can occur between 3 cells or sometimes more [78]. Entotic cell engulfment led to damage in ECM like anoikis; however, their functional process is different. Entosis is one of the cell's cannibalistic behaviors, killing the neighbor cells with the help of the E-cadherin receptor [79]. There has been no direct research approach to link entosis with SCI. However, entosis have been observed when embryonic stem cells have been cultured with mesenchymal stem cells [80]. So, it may hamper stem cell-based spinal regeneration approaches.
Methuosis is nonapoptotic cell death; the mechanism of cell death is related to cytoplasmic fluid displacement by large vacuoles generated by macropinosomes. Rab5 and Rab7 GTPase proteins are involved in the transportation of vacuoles. There is currently no evidence to link methuosis with SCI. However, methamphetamine leads to the death of the neurons in the central nervous system via methuosis [81]. Entosis and methuosis are the new domain for SCI that researchers can approach to bring the novel idea to avoid neuronal damage during SCI.
2) Mitochondria-mediated
(1) Mitoptosis in SCI
Mitochondria is an essential organ to produce energy for cell division and homeostasis. Mitoptosis, also known as mitochondrial suicide, occurs when the mitochondria divide and fuse, cutting off the adenosine triphosphate (ATP) supply and leading to apoptosis and autophagy [82,83]. As a result, they transform into autophagosomes or mitoptotic entities and are ejected. Hence, mitoptosis is a mitochondrial death mechanism rather than a cell death mechanism. However, the high fission or fusion breaks mitochondria apart, ultimately leading to cell death [84]. When BAK/BAX permeabilize the outer mitochondrial membrane proteins, they release a protein called deafness-dystonia peptide (DDP), also known as translocase of inner mitochondrial membrane 8a (TIMM8a) that attaches to cytoplasmic DRP1 to bring DRP1 into the mitochondria to promote mitochondrial fission [85]. A thorough analysis using electron microscopy, as well as the imaging of fragmented mitochondria with mitochondria-specific dyes (MitoTracker Green, Thermo Fisher Scientific Inc., Waltham, MA, USA) using fluorescence microscopy, may offer information about the existence of mitoptosis. In addition, antibodies targeting TIMM8a/DDP and cytochrome-c are also used [85,86]. Mitoptosis occurs due to changes in the membrane of the mitochondria, membrane condensation with swelling and fragmentation in cristae [83].
Mitoptosis is observed in SCI triggered by Ca2+ accumulation in the cellular matrix by glutamate excitotoxicity [86]. To investigate a putative mitochondrial-SCI relationship, Wingrave et al. [87] produced a 40 g/cm force injury in rats by contusion, and 4 hours, 1-cm slices of spinal cord tissue were collected for calcium green (2-AM) staining, western blot, and immunohistochemistry. The penumbra and lesion tissue sections showed free intracellular calcium (Ca2+) levels increased following the injury compared to sham-operated (control) rats. After SCI, the mitochondria-mediated cell death pathway was activated in the penumbra and lesion by elevating Bax: Bcl-2 ratio via western blot. Wei et al. observed neural damage attenuation and locomotor function improvement in rats [88].
(2) Parthanatos in SCI
It is a type of caspase-independent PCD activated upon specific types of DNA due to the hyperactivation of the DNA repair gene poly ADP-ribose polymerase 1 (PARP1) [89]. Poly-ADP ribose is (PAR) produced by PARP1 that translocates from the nucleus to the cytoplasm, interacting with mitochondrial proteins and releasing apoptosis-inducing factors (AIFs). Then it couples with the macrophage migration inhibitory factor (MIF). This MIF: AIF complex condenses chromatin and breaks DNA in the nucleus [90]. Unlike apoptosis, intact PARP and its activation are required to initiate parthonatos instead of PARP breakdown. Parthanatos is also unaffected by broad-spectrum caspase inhibitors [91].
Moreover, unlike apoptosis, DNA fragmentation is considerable [92]. Biomarkers for parthanatos include nuclear AIF, PARP-1 activation, and PAR accumulation. Mitochondrial depolarization may be detected using fluorescent probe labelling, proving that the method works [93]. Yang et al. [94] observed by interrupting MIF: AIF interaction by knocking down MIF-provided neuroprotection from oxidative stress-induced parthanatos post-SCI. Secondary damage after an initial traumatic or nontraumatic injury is the primary concern of parthanatos in SCI [95]. In post-SCI damage, metabolic disruption and glutamine excitotoxicity are the 2 frequent biochemical pathways [96]. Before the word pathanatos, glutamine excitotoxicity was widely studied concerning cell death [97]. In vitro and in vivo glutamine excitotoxicity models were developed using kainate, a glutamine analogue. In vitro, kainate-induced neuron death was primarily mediated by parthanatos than apoptosis [98]. The inhibitor of PARP-1, 6-5(H)-phenanthridine (PHE), prevented AIF translocation and overactivation of PARP-1, both of which are associated with parthanatos [99].
PJ34, another PARP-1 inhibitor, reduced kainite excitotoxicity. In kainate-treated mice, PJ34 and PHE had minor impacts on locomotor network damage, showing that some areas of the spinal cord may be resistant to parthanatos. Parthanatos killed most neurons, making them more susceptible to excitotoxicity after SCI. However, glial cells were more resistant to excitotoxicity and perished mainly via apoptosis [100]. Oxidative stress contributes to subsequent SCI damage. The inhibition by a polyadenosine diphosphate-ribose polymerase-1 (PARP-1) inhibitor 3-amniobenzamide may help [96]. Parthanatos has been linked to cell death caused by oxidative stress after damage. In the presence of Mg2+ in a pathological medium simulating metabolic disruption after ischemic SCI in vitro, parthanatos was detected in spinal white matter and partial portions of spinal grey matter [101]. JNK1 and JNK3 have also been linked to parthanatos premitochondrial activation [102].
3) Iron-mediated
(1) Ferroptosis in SCI
The term “Ferroptosis” was coined in 2012 as a nonapoptotic PCD triggered by iron accumulation inside the cell [103]. In ferroptosis, the cell maintains a normal-looking morphology and a normal-sized nucleus devoid of chromatin condensation. System XC- is an amino acid antiporter that helps move l-glutamate inside cells and l-cystine outside cells across the plasma membrane of cells contributing to various human processes [103]. A defect in glutathione peroxidase 4 (GPX4) or system XC- causes a collapse in glutathione-dependent antioxidant defense. For glutathione production, system XC- carries cystine into the cell and converts it to cysteine from the extracellular cystine. To reduce cellular lipid peroxidation, GPX4 may directly catalyze glutathione-lipid hydroperoxide interactions. Leaky gut and GPX4 suppression lead to the accumulation of lipid hydroperoxide that reacts with free iron to form lipid reactive oxygen species (ROS) leading to the death of the cell [104]. It is distinguished from apoptosis by morphology, biochemistry, and genetics [105]. In ferroptosis, ROS grows iron-dependently and has a vital role [106]. An upsurge in the number of studies on ferroptosis in SCI has occurred in the last few years. Ferroptosis has been related to excitotoxicity-induced cell death. GPX4 reduces ferroptosis by promoting motor neuron health and survival. So, ferroptosis suppression by GPX4 is essential [104]. Deferoxamine, a drug of iron toxicity, has been shown in previous studies to lower total Fe2+ ions, caspase-3, IL-1β, and TNF-α expression levels after SCI and inhibit the creation of glial scars and apoptosis from increasing function recovery [107]. Another study on the effect of proanthocyanidins on SCI repair indicated that intraperitoneal injections of proanthocyanidins suppressed ferroptosis, which improved functional recovery after SCI [108].
Ferroptosis may have a substantial role in secondary damage after SCI, and inhibiting this process benefits recovery after SCI, according to these studies. On the other hand, adequate research on the role of ferroptosis in SCI still needs to be done. Research is urgently needed to establish the function of ferroptosis, which may lead to new therapeutic options for SCI in glial scar formation and neuronal death [109].
Copper can induce cell death, also termed cuproptosis. FDX1-mediated mitochondrial proteotoxic stress causes cuproptosis. FDX1 converts Cu2+ to Cu+, boosting lipoylation and aggregation of mitochondrial TCA cycle enzymes (particularly DLAT). In addition, FDX1 inactivates Fe-S cluster proteins. Cu importers (SLC31A1) and exporters (ATP7B) change intracellular Cu+ levels to impact cuproptosis sensitivity [110]. Even though it has not been linked with SCI, Enge et al. [111] found elevated Cu concentrations in skeletal muscle and the spinal cord in the presymptomatic stage, which worsened with disease development in the SOD1G93A-mutant mice amyotrophic lateral sclerosis model.
4) Immune factors mediated
(1) Pyroptosis in SCI
Pyroptosis is a nonapoptotic PCD that happens in immune cells as a response when intracellular pathogens release inflammatory signals. Infected macrophages’ inflammatory sensors, like NOD-like receptors (NLRs), detect flagellin molecules in pathogens and induce the development of multiprotein complex inflammasomes, which then activate caspase1 [112]. When activated, caspase1 causes membrane hole formation by cleaving gasdermin D, causing the cell membrane to tear [113]. DNA condensation and fragmentation are also seen throughout the process. Furthermore, bacterial lipopolysaccharide (LPS) directly activates caspase11, causing pyroptosis [114]. Pyroptosis can be determined through gasdermin D cleavage through western blot analysis, IL-1 through caspase activation, and visualization of membrane integrity loss through fluorescence microscopy and the quantification of released cytoplasmic lactate dehydrogenase [115].
Epithelial cells, neurons, and pyroptotic keratinocytes, as well as myeloid-derived professional phagocytes such as DCs and macrophages, all, have been identified to show pyroptosis. In addition, pyroptosis has been linked to antibacterial and inflammatory responses during infection [116].
Inflammasomes containing the caspase-1 enzyme have been found to inhibit pyroptosis in the amygdala kindling model of neurological illness [117]. Silencing Nucleotide-binding oligomerization domain-1 (NLRP1) or caspase-1 also decreased pyroptosis in rats [118]. Deficient microglial voltage-gated proton channels may inhibit NLRP3-induced neuronal pyroptosis. Sevoflurane-induced neuronal pyroptosis is connected to the Bach1/Nrf2/Erk1 signaling pathway [119]. Streptococcus pneumoniae can cause pyroptosis in murine microglia, requiring the NLRP3 inflammasome, which activates caspase-1 [120]. The function of pyroptosis in SCI pathogenesis is unknown. Pyroptosis and the inflammatory response to SCI should be explored further.
(2) Netosis in SCI
Netosis is a nonapoptotic PCD caused by pathogenic infections or their components. It is most frequent in immune cells, notably neutrophils. It causes the nucleus or mitochondria of neutrophils to generate extracellular traps (NETs) composed of modified chromatin-coated neutrophil DNA and bactericidal proteins from granules and cytoplasm [121]. The most common type is suicidal netosis, where neutrophils die after releasing NETs [122]. When neutrophils recognize pathogen-associated molecular pattern signals from pathogens, that activates the mitogen-activated protein kinase complex to produce protein kinase (PKC). PKC activates NADPH oxidase, causing ROS generation. Increased Ca2+ levels in mitochondria cause mitochondrial permeability transition pore (mPTP) to open and release mitochondrial ROS (mtROS) [123]. Mitochondrial dynamin like GTPase (OPA1) ensures the production of NAD+, which is eventually converted to nicotinamide adenine dinucleotide (NADH) through glycolysis. Lastly, NADH transports electrons in the mitochondrial electron transport complex, which upon activation, supplies ATP for NET synthesis [124].
ROS with mtROS causes azurosome release by rupturing azurophil granules. Neutrophil elastase (NE) and myeloperoxidase (MPO) release and translocates along with a gene named Peptidyl arginine deiminase 4 (PADI4) in the nucleus [125]. MPO generates HOCl- to start secondary injury progression, and NEs cleave the Gasdermin D pore at the nuclear membrane to release NETs [126]. PADI4 enters the nucleus and causes chromatin decondensation by citrullinating histones (H3, H2A, and H4) and inhibiting glutaredoxin 1 to disrupt cytoskeletal dynamics of the membrane [127]. Another type of netosis, also known as vital netosis, is where the neutrophil does not die. It is believed to occur by the NET formation of mitochondrial DNA rather than the nuclear DNA of neutrophils. It occurs by the Activation of Toll-like receptor 4 (TLR4) and is regulated by small conductance calcium-activated potassium (SK3) channels [128]. Elevated cytosolic Ca2+ increases in SCI pathology promote complex 1 activity and ATP and ROS generation [129]. Tissue damage, Hypoxia, oedema, and tissue damage cause the synthesis of the first soluble mediator of ROS [130]. The role of ROS in developing networked systems is significant. Because of this, SCI may induce NETosis. The mitochondrial calcium uniporter also allows Ca2+ into the mitochondria. Ca2+ levels inside cells may promote membrane permeability. NADPH Oxidase activation or other nonoxidative mechanisms connected to mPTP. Thus, NETs develop throughout the SCI process. Neutrophils infiltrate the spinal cord following SCI [131]. These cytotoxic elements cause more extensive lesions and decrease neurological function. MPO increases neutrophil infiltration, causing secondary injury and slowing SCI recovery [132].
Swelling oedema in the spinal cord astrocytes may cause poor functional recovery and treatment resistance following SCI. Rat spinal cord astrocytic oedema was reduced when oxygen-glucose deprivation and reperfusion (OGD/R) were combined with the high mobility group box protein 1 (HMGB1) shRNA or ethyl-pyruvate treatment. Conversely, when OGD/R was applied to spinal cord astrocytes, HMGB1 increased aquaporin-4 (AQP4) expression and cell swelling through HMGB1/TLR4/nuclear factor-kappa B (NF-κB) signaling. Inhibitors of TLR4 and NF-κB have also been demonstrated to reduce activation effects [133]. TLR4 induces NETosis and increases damage after cerebral ischemia and thrombosis [134]. Therefore, NET development may affect SCI damage. However, no proof exists. However, some research suggests neutrophils may benefit tissue regeneration in SCI since their inflammatory activity may promote healing [135]. Various unanswered concerns surround neutrophil activity in SCI. To get new insights into traumatic brain damage, further study of their cellular and molecular pathways is necessary (SCI).
5) Other factors mediated
(1) Necroptosis in SCI
Also known as programmed necrosis, it activates receptorinteracting protein kinases (RIPKs) by Activation and crosstalk through many signaling pathways. RIPKs are triggered by the employment of several cell surface receptors to macromolecular complexes: T-cell receptors, DRs, and TLRs. RIPK1 and RIPK3 are essential components of the necrosome. RIPK3 phosphorylates the downstream molecule mixed lineage kinase domainlike protein (MLKL), causing MLKL oligomerization. An oligomerized MLKL enters the cell membrane and permeabilizes it, causing cell death [136]. Furthermore, regulatory factors of DNA-dependent activators of interferon synthesized after viral infestation, double-stranded viral DNA, and cytosolic DNA sensors activate RIP3-dependent necroptosis. It exhibits necrotic morphology, including membrane rupture and organelle loss. An increasing amount of research shows that, unlike necrosis, necroptosis is caspase-independent [137]. It is relevant and well established in ischemic brain damage, viral myocardial infarction, and neurodegenerative diseases [138]. The most well-studied necroptosis pathway is TNF-R1 binding to TNF [139]. Research suggests necroptosis is involved in an intracellular signaling cascade involving MLKL and RIP1/3 kinase [136]. RIP1 kinase activity is required for necroptosis activation. Holler et al. [140] discovered that proteasome subunit beta type-4 controls the RIP3 and MLKL pathways using TNF-induced necroptosis cell culture. Wang et al. [141] discovered necrostatin-1 (NEC-1), a novel small molecule suppresses necroptosis by modulation of the formation of protein complexes of RIPK1 and RIPK3 and by recruiting RIP1/3–MLKL. After SCI, Nec-1 improved the histopathology and functional impairments, indicating that necroptosis may contribute to brain cell death. Thus, Nec-1 may be used to treat SCI [141].
Increasing evidence links SCI necroptosis to inflammation. The ER of necroptotic microglia/macrophages can modulate SCI-induced inflammation [142]. Fan et al. [143] discovered that M1 macrophages/microglia might induce necroptosis in reactive astrocytes through the MyD88/TLR signaling pathway. Smad ubiquitination regulatory factor-1 (Smurf1) may promote neuron necroptosis following LPS-induced neuroinflammation, suggesting it might be a therapeutic target [144]. After spinal cord damage, necroptosis-induced glial and neuronal cell death has been reported. Activating necroptosis in the CNS may cause cell death and tissue damage. Despite substantial research into necroptosis after SCI, our knowledge is limited. Secondary damage can only be addressed if necroptosis is linked to SCI etiology [145].
(2) Other minor pathways in SCI
Other minor pathways may also lead to SCI progression. Oxieptosis is a novel nonapoptotic PCD that is triggered by the accumulation of ROS. Kelch-like ECH-associated protein 1 (KEAP1) C-terminal cysteines are oxidized by moderate ROS levels, causing degradation of the KEAP1-NRF2 complex and nuclear translocation of NRF2 [146]. Antioxidant genes are produced by Nrf2 that help to remove ROS from the nucleus. High amounts of intracellular ROS trigger the production of phosphoglycerate mutase family member 5 by KEAP1, which dephosphorylates apoptosis-inducing factor 1 (AIFM1) at the Ser116 position after binding to it, ultimately leading to this kind of cell death [147]. ROS significantly affects the primary and secondary injury progression for SCI [148]. Due to ischemic injury at the primary injury site, free ROS gets generated, which links with the disruption in Nrf2 translocation and decreasing neuronal pH and form of mPTP complex [72,149]. So even though there is no current research linking oxieptosis to SCI, it is a starting point to start many cell death pathways relevant to SCI. So, it is a hot topic for researchers to explore.
pH has a significant role in the progression of cell death in SCI. Alkaliptosis is a new pH-dependent cell death relevant to many diseases [150]. Tang et al. [151] define alkaliptosis as pH-mediated, N-acetyl cysteine-mediated, drug-induced cell death. Scientists have established that alkalinization of the cellular milieu with sodium hydroxide promotes cell death using NF-kB. Unlike ferroptosis and necrosis, alkaliptosis is a chemical process. NF-kB activation may prevent alkaliptosis [152]. The Nrf2/HO-1 signaling pathway may activate proinflammatory and apoptosis-inhibitory genes. Over or underexpression of the Nrf2/HO-1 signaling pathway has antiapoptotic effects that can be utilized against this type of cell. Currently, there is no research done to link alkaliptosis with SCI. Furlong et al. [153] showed that intracellular acidification induces apoptosis by stimulating IL-1β converting enzyme like protease activity. In the isolated spinal cord, Jalalvand et al. [154] showed that both increased and decreased pH reduced the locomotor burst rate, proving a possible link between alkaliptosis and SCI. Treatment strategies can be built around it as it heavily depends upon the NF-kB pathway [155].
Autoschizis is another bizarre type of cell death where the cell develops cracks. These cracks develop inside the cell organism, followed by getting destroyed by proteases that also develop inside the cell. The standard cell remains unaffected, but the cracked cells die off [156]. There is currently no link between autoschizis and SCI. However, it is a potent necrosome trigger that may contribute to the necrosis and necroptosis progression in SCI [157].
3. Nonprogrammed Nonapoptotic Cell Death
1) Nonprogrammed necrosis in SCI
Necrosis is a nonprogrammed type of cell death triggered by toxins, physical injuries, and infections disrupting ionic pumps leading to Ca2+ influx, resulting in morphological alterations such as cytoplasmic swelling (oncosis), consequential intracellular organelle loss with little to no chromatin condensation, and plasma membrane rupture [158]. It starts with the acute phase comprising a variety of mechanisms, including ionic imbalance and glutamate excitotoxicity, toxic blood component buildup, the release of proinflammatory cytokine by lymphocytes and neutrophils, ATP depletion, and free radical production [159]. As the damage continues, in the subacute phase, surviving axonal demyelination, neuronal apoptosis, axonal bulb retraction (dieback), Wallerian degeneration, glial scar formation surrounding the injury site, and matrix remodeling occur. In the chronic damage phase, further changes occur, such as maturation of the glial scar, increasing axonal retraction bulb (dieback), and creating a cystic cavity [160]. It is common in trauma, ischemia, and potentially certain kinds of neurodegenerative conditions. It is typically regarded as a passive process that requires very little energy yet does not need de novo macro molecular synthesis [161]. The process of cell death is a continuum of apoptosis and necrosis. Variable levels of crosstalk harmony between coexisting apoptotic and necrotic processes might contribute to neuronal death along this spectrum [162]. The contribution of necrosis in SCI pathology has been well established in both in vitro and in vivo models of acute and chronic SCI [163]. Acute SCI results in necrosis, known as progressive hemorrhagic necrosis. It is a poorly understood pathological process marked by necrosis and bleeding that results in severe cystic cavitation of the spinal cord, profound neurological impairment, and spinal cord tissue loss [164]. A necrosis initiator gene, TNF-α, produces death in oligodendrocytes in the spinal and supraspinal region [165]. Its antagonist significantly reduced oligodendroglial necrosis in SCI [166]. Excessive N-methyl-D-aspartate receptor activation in neurons during glutamate-induced excitotoxicity may result in necrotic cell death and progression of secondary injury [167]. These secondary processes contribute to the evolution of pathological abnormalities in severe injuries, from central hemorrhagic necrosis involving predominantly grey matter to infarction of both white and grey matter proximally and distally at the injury site. Less severe damage changes axons and myelin [168] (Table 1).
CONCLUSION AND FUTURE PERSPECTIVE
The ensuing cascade of cell death after CNS injury or ischemia has long been considered a target for neuroprotective drugs to preserve tissue and function. Defects in one or more cell death processes are connected to various spinal cord injuries, including neurodegenerative conditions involving abnormal cell destruction. The regulators and signaling pathways of the various cell death mechanisms continue to be appealing therapeutic targets that have the potential to serve as the foundation for translational research that may result in improvements for patients afflicted with these disorders. As a concluding statement, we can say that a significant amount of additional research, including both fundamental works in studies of animal models involving clinical trials, is required to acquire a more in-depth knowledge of the various processes modulating cell death in SCI. Many cell death pathways for axonal/neuronal regeneration and proinflammatory signaling-induced secondary injury in SCI are undiscovered. This review thus expands the extent of the cell death pathways in the SCI, and knowledge must be used to make revolutionary advancements in treating these illnesses.