Primary Spinal Cord Oligodendroglioma with Postoperative Adjuvant Radiotherapy: A Case Report
Article information
Abstract
Primary spinal cord oligodendrogliomas are rare tumors comprising two percent of all spinal cord tumors. Although a treatment guideline has yet to be established, maximal surgical resection is primary in the treatment of spinal cord oligodendrogliomas. Adjuvant radiotherapy has remained controversial, and it is unclear whether chemotherapy adds any benefit. In this case report, the authors present a 24-year-old male who had a seven-year history of left leg weakness and a radiating pain in both legs. Magnetic resonance image (MRI) showed an intramedullary mass at the T4-T8 level. He underwent subtotal removal of the tumor and pathologic diagnosis revealed a WHO grade II oligodendroglioma. The patient was treated with radiotherapy postoperatively and followed up with MRI annually. Clinical and radiological status of the patient had been stationary for four years after the surgery. The five-year follow-up MRI showed an increase in the size and extent of the residual tumor. Despite radiological progression, considering that symptoms and the performance status of the patient had remained unchanged, further treatment has not been performed. Given the clinical outcome of this patient, close observation after subtotal removal with adjuvant radiotherapy is one of the acceptable treatment options for WHO grade II spinal cord oligodendrogliomas.
INTRODUCTION
Primary spinal cord oligodendrogliomas are very rare tumors, comprising only two percent of all spinal cord tumors2). The overall prognosis with regard to oligodendrogliomas of spinal cord is apparently worse than that of low-grade astrocytomas4). For intracranial WHO grade II oligodendrogliomas, maximal safe surgical resection followed by adjuvant radiotherapy is the standard treatment711). However, in the case of spinal cord oligodendrogliomas, there is no uniform treatment guideline. Maximal safe surgical resection of the tumor is the standard treatment, but adjuvant radiotherapy has remained controversial4). In the present case, we report a patient with WHO grade II spinal cord oligodendroglioma who underwent subtotal tumor removal and adjuvant radiotherapy with good functional outcome and disease control.
CASE REPORT
1. History
A 29-year-old male presented with a seven-year history of left leg weakness and a radiating pain in the both legs. His past medical and family history was unremarkable. He visited a local primary clinic and was diagnosed with a herniated lumbar intervertebral disc. Physical therapy was performed but was not effective. As his symptoms had worsened gradually, in 2007, he was referred to a local neurosurgery clinic. Cervical and thoracic magnetic resonance image (MRI) was performed and revealed a fusiform weakly enhancing mass ranging from the T4 to T8 level with associated hydrosyrinx. He was referred to the neurosurgery department at our institution.
2. Examination
A physical examination revealed muscle weakness of grade 4/5 in the left lower extremity, which resulted in a mild gait disturbance, a radiating pain in the bilateral lower extremity, hypesthesia below the umbilical level, deep tendon hyperreflexia of the bilateral ankle and knee, and bilateral ankle clonus. No urinary symptoms were observed. The MRI performed outside showed a large intramedullary fusiform mass ranging from the T4 to T8 level with associated hydrosyrinx ranging from the T2 to T4 level. The mass was isointense on T1-weighted images (WI) (Fig. 1A) and slightly hyperintense on T2WI with a relatively good demarcated margin (Fig. 1C). On postgadolinium images, focal weak heterogeneous enhancement was noted at the upper part of the tumor (Fig. 1B). Computed tomography (CT) was also performed and showed calcification in the center of the tumor (Fig. 1D).
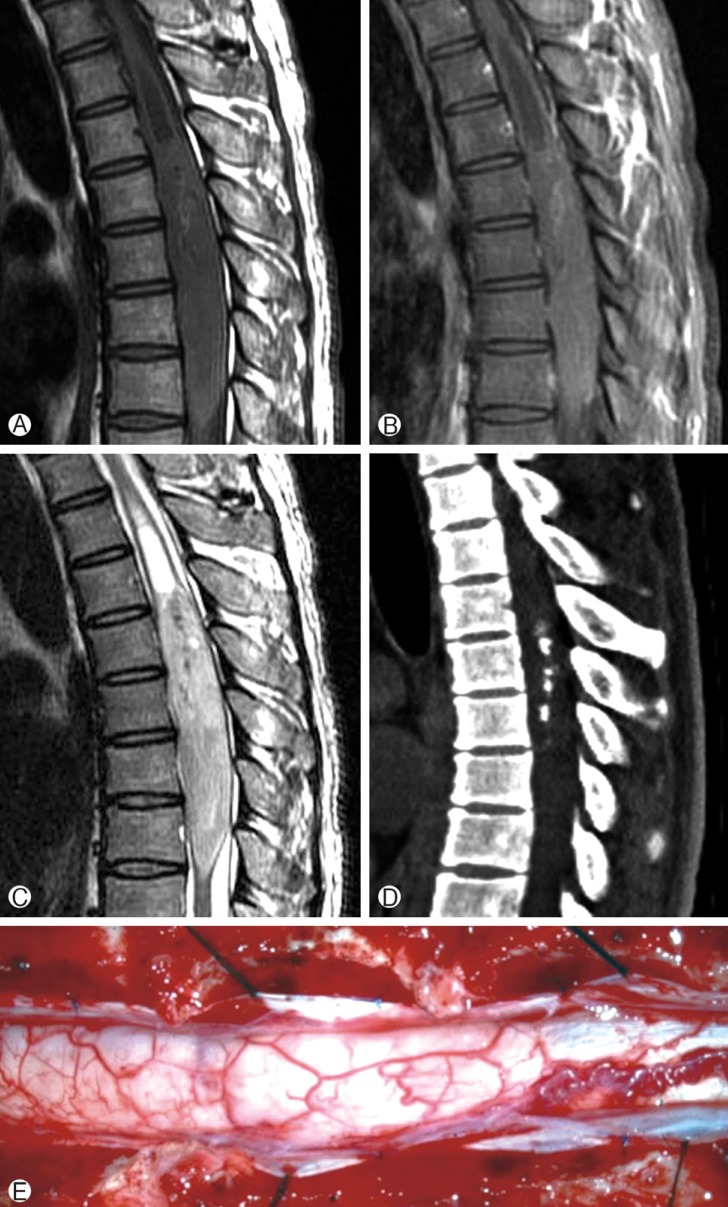
Sagittal T1-weighted without contrast (A), T1-weighted with contrast (B), and T2-weighted (C) magnetic resonance image (MRI) of the thoracic spine showing an intramedullary heterogeneous iso T1, high T2, and heterogeneously enhancing lesion located at the T4-T8 level associated with the syrinx extending from T2 to T4. Sagittal computed tomography (CT) images of the thoracic spine without contrast showing focal calcification in the center of the spinal cord tumor (D). Intraoperative photograph showing an edematous and hypervascularized spinal cord with midline effacement (E).
3. Operation
The patient underwent a T3-T9 laminectomy. Intraoperatively, a tense dura was found. After vertical incision of the dura, an edematous and hypervascularized spinal cord was exposed with midline effacement (Fig. 1E). Midline myelotomy from T3 to T8 was performed with an ophthalmic knife, scissors and dissectors, which revealed grayish, gelatinous and firm tumor tissue. The hydrosyrinx was opened during an exploration of the T3-level spinal cord, which resulted in a sunken spinal cord below the T4 level. A plane between the spinal cord and the tumor was developed from the superior aspect of the tumor at the T4 level using microsurgical techniques. The tumor was difficult to distinguish from the cord, and the plane was indistinct. An intraoperative frozen tissue biopsy led to a diagnosis of a low-grade glioma rather than a gangliogenic tumor or an ependymoma. Due to poor demarcation between the tumor and the spinal cord, the tumor was subtotally removed by internal debulking using a Cavitron ultrasonic surgical aspirator. Meticulous hemostasis was accomplished. Without closure of the pial layer, dura edges were approximated with a watertight closure with an artificial dura. After saline irrigation, the surgical wound was closed in anatomical layers.
4. Histological Findings
The histopathological examination represented a highly cellular tumor with dysmorphic calcification, prominent hyalinized blood vessels and Rosenthal fibers in the periphery of the tumor. There was no vascular endothelium hypoplasia or necrosis. The permanent sections of this tumor showed the classic pattern of low cellular monotonous round cells with a clear cytoplasm upon hematoxylin and eosin (H&E) staining (Fig 2A). The immunohistochemistry of the glial fibrillary acidic protein (GFAP) revealed that most cells were negative, with some positive reactive astrocytes (Fig 2B). Other immunohistochemistry stains for Galectin-3, Neu-N, synaptophysin and CD99 were positive, while p53, EGFR and neuronspecific enolase (NSE) were negative. The Ki-67 labeling index was less than 1%(Fig 2C). A molecular pathologic examination by fluorescence in situ hybridization (FISH) revealed no codeletion of chromosomes 1p and 19q. Another molecular examination of O6-methylguanine-DNA methyltransferase (MGMT) gene promoter methylation by polymerase chain reaction (PCR) was performed and was found to be positive. The final pathological diagnosis was a low-grade glioma, most likely a WHO grade II oligodendroglioma.

Histological findings of resected specimens. (A) Hematoxylin and eosin (H&E) staining shows low cellular monotonous round cells with clear cytoplasm(H&E, ×200). (B) Upon glial fibrillary acidic protein immunostaining (GFAP), most cells are negative with some GFAP positive reactive astrocytes (GFAP immunostaining, ×200). (C) The Ki67 labeling index is less than 1% (Ki67 immunostaining, ×200).
5. Postoperative Course
Immediate postoperatively, the motor power of the lower limbs remained unchanged at grade 4/5, and paresthesia was improved slightly. However, hypesthesia was newly developed with worsened spasticity and hyperreflexia. With a thoracolumbosacral orthosis, the patient was able to walk with a cane. After two weeks of rehabilitation, the patient recovered to his preoperative condition and was discharged. The residual tumor was treated with radical radiotherapy at 45Gy administered in 25 fractions over five weeks, starting one month after the operation. Since the adjuvant radiotherapy, MRI has been performed annually, showing no significant interval change in the size, extent and enhancement pattern of the residual tumor during the initial four-year postoperative period. A postoperative five-year follow up MRI revealed an increase in the size and extent of the residual tumor with cystic change and focal gadolinium enhancement (Fig. 3). In spite of the radiological progression of the tumor, there were no changes in symptoms or performance status. Therefore, the patient is on close observation with follow up through MRI annually without any additional treatment.

Sagittal T1-weighted with contrast-enhanced MRI of one-year follow up (A), two-year follow up (B), three-year follow up (C), four-year follow up (D), and five-year follow up (E).
DISCUSSION
Primary oligodendrogliomas of the spinal cord are rare and represent 2% of all tumors arising from the spinal cord and film terminal and 1.59% of all oligodendrogliomas23). Since the first report of a primary spinal oligodendroglioma by Kernohan et al.8) in 1931, including this case report only 56 cases of primary spinal cord oligodendrogliomas have been reported in the literature23468913).
Primary oligodendrogliomas occur in both children and adults, with a mean age of 28.4 years old according to the literature review by Fountas et al.4). It is widely accepted that intracranial oligodendrogliomas are more common among males; however, the existent literature shows that there is no significant gender trend with regard to spinal oligodendrogliomas14). By the literature, most commonly presenting symptoms are spinal pain and sensory deficit, followed by motor deficit and voiding problems, which depend on the anatomic location of the tumor. Occasionally, several case reports have revealed increased intracranial hypertension due to diffuse oligodendrogliomatosis6). The most common anatomic locations of spinal oligodendrogliomas are the thoracic level, followed by the cervical, thoracocervical junction, lumbar and thoracolumbar levels47).
MRI is the diagnostic method of choice. Primary spinal oligodendrogliomas usually appear as heterogeneous hypoor iso-signal intense lesions on T1WI and as hyper-signal intense lesions on T2WI14). The vast majority of spinal oligodendrogliomas demonstrate mild to moderate heterogeneous spotty enhancement on postgadolinium images14). Cystic components or cystic necrotic areas may be observable, particularly in cases of high-grade spinal oligodendrogliomas5714). In addition to MRI, plain X-rays and CT scans can be helpful for evaluating tumor-associated skeletal deformities and instances of calcification. The presence of calcification in spinal cord oligodendrogliomas has been estimated to range from 28% to 40%1). In this case, the initial MRI showed typical iso-intense lesions on T1WI and hyper-intense lesions on T2WI, with focal heterogeneous weak enhancement. The margin of the tumor was relatively well demarcated by MRI; however, the tumor was difficult to distinguish from the normal spinal cord intraoperatively. A CT scan showed calcification in the tumor. Postoperative five-year follow-up MRI revealed an increase in lesions size and extent of the residual tumors with cystic changes and focal enhancement, suggesting the possibility of a highgrade component as well as changes related to the radiotherapy.
The classic microscopic appearance of oligodendrogliomas is an infiltrating glial neoplasm composed of monotonous clear cells with perinuclear halos, called a "fried egg" appearance, embedded in a scaffolding of "chicken wire" vessels14). According to immunohistochemical analyses, spinal oligodendrogliomas generally show no expression to GFAP but occasionally reveal focal immunoreactivity due to the presence of scattered astrocytes in the tumor bed or the presence of minigemistocytes147). In adult intracranial oligodendrogliomas, approximately 80% contain characteristic chromosome 1p36 and 19q13 co-deletion, which appears to correlate with chemosensitivity7). In this case, immunohistochemistry on GFAP was focal-positive, which are compatible with minigemistocytic oligodendrocytes. Interestingly, there was no chromosome 1p/19q co-deletion according to FISH in this case. This is one of the reasons that radiotherapy was preferred over chemotherapy as an adjuvant therapy for this patient. Another molecular pathologic examination of MGMT gene-promoter methylation by PCR showed a positive result. According to a literature review done by the authors, this is the first case report of the primary spinal oligodendroglioma with MGMT gene-promoter methylation and without 1p/19q co-deletion. More case reports and long-term follow up are necessary to understand the clinical significance of these results.
There is no uniform treatment guideline, but of key importance during treatment is maximal surgical resection of the tumor37). However, despite the use of intraoperative electrophysiological monitoring and microsurgical techniques, surgical removal of spinal cord oligodendrogliomas is challenging in many cases due to the infiltrative characteristic of the tumors and high-grade malignancy in a large proportion of reported cases13461012). Interestingly, among the cases reported in the literature, gross total resection was achieved in only 8 out of 56 cases47). Although this patient underwent surgery with intraoperative electrophysiological monitoring and microdissection techniques, a poorly demarcated tumor margin prevented radical resection of the tumor.
The effectiveness of adjuvant radiotherapy is still controversial4). Although Fortuna et al.3) in 1980 reported that radiotherapy after an operation appeared to prolong survival; their study was limited in its evaluation given its analysis of only eleven cases prior to 1980. On the other hand, radiotherapy has been linked to complications such as post-radiation myelopathy and radiation-induced vertebral column deformities, especially among pediatric patients312). Therefore, the decision to use postoperative radiotherapy should be made very carefully, considering the age, extent of resection, grade and histologic characteristics of the tumor, as well as the genetic characteristics of the tumor7). In the present case, conditions such as a young age, a large residual WHO grade II oligodendroglioma and negative results for 1p/19q co-deletion were good indications for postoperative adjuvant radiotherapy. After radiotherapy, there was no evidence of the radiation induced complications described above until the fifth year postoperative follow-up examination.
Postoperative chemotherapy has been employed in limited cases of partially resected spinal oligodendrogliomas15610). McLendon et al.1) and Ramirez et al.10) reported spinal oligodendroglioma patients treated with procarbazine, lomustine, and vincristine. Guppy et al.6) reported a good response after administering temozolomide with concurrent local radiotherapy of 45Gy for a spinal oligodendroglioma with co-deletion of 1p and 19q. However, these data are very limited, and chemotherapy has been considered as the only adjuvant treatment option for cases of partially resected high-grade oligodendrogliomas for which radiotherapy may have severe side effects7). For the current case, there was only MGMT gene promoter methylation without chromosome 1p/19q co-deletion, which is not favorable for chemotherapy. However, given the revealed radiological progression of the tumor after postoperative radiotherapy, additional chemotherapy can be considered as a further treatment option.
The postoperative prognosis of spinal oligodendrogliomas has been reported to be less favorable than that of low-grade intramedullary astrocytomas3). Fortuna et al.3) reported in their review that the mean survival time was 28.6 months. In this study, we report a spinal cord oligodendroglioma patient with four years of progression-free survival, only after subtotal removal of the tumor and adjuvant radiotherapy.
Despite the radiological progression of the disease, and considering that the symptoms and performance status of the patient remained unchanged, further treatment has not been performed and the patient is on close observation with annual MRI follow ups. If the performance status of the patient is aggravated or radiological progression become evident, a second operation or adjuvant chemotherapy should be considered.
CONCLUSION
Spinal cord primary oligodendrogliomas are rare tumors without a standard treatment guideline. Aggressive tumor removal is the most important step, but it is challenging to obtain maximal surgical resection due to the infiltrative nature of these tumors and risk of postoperative neurological deficit. Adjuvant radiotherapy and chemotherapy remain controversial. In this case, the patient underwent subtotal tumor removal followed by adjuvant radiotherapy without treatment-related complications. During postoperative five-year follow ups without any additional treatments, the clinical status of the patient has been exceptionally stationary, only with recent radiological progression of the tumor according to an MRI exam. Therefore, we concluded that given the lack of an established treatment guideline, maximal safe resection with adjuvant radiotherapy is an acceptable treatment option for primary spinal cord WHO grade II oligodendrogliomas.
ACKNOWLEDGEMENT
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning (2014R1A2A1A11049662).
This work was also supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (No. 2010-0028631).