Glioma Immunotherapy: Advances and Challenges for Spinal Cord Gliomas
Article information

Abstract
Spinal cord gliomas are rare entities that often have limited surgical options. Immunotherapy has shown promise in intracranial gliomas with some research suggesting benefit for spinal cord gliomas. A focused review of immunotherapies that have been investigated in spinal cord gliomas was performed. The primary methods of immunotherapy investigated in spinal cord gliomas include immune checkpoint inhibitors, adoptive T-cell therapies, and vaccine strategies. There are innumerable challenges that must be overcome to effectively apply immunotherapeutic strategies to the spinal cord gliomas including low incidence, few antigenic targets, the blood spinal cord barrier, the immunosuppressive tumor microenvironment and neurotoxic treatment effects. Nonetheless, research has suggested ways to overcome these challenges and treatments have been effective in case reports for metastatic non-small cell lung cancer, melanoma, midline glioma and glioblastoma. Current therapies for spinal cord gliomas are markedly limited. Further research is needed to determine if the success of immunotherapy for intracranial gliomas can be effectively applied to these unique tumors.
INTRODUCTION
Primary tumors of the spinal cord comprise a rare 4.5% of all central nervous system (CNS) tumors [1]. A large proportion of these are gliomas which can be further subclassified based on cellular origin into ependymomas, astrocytomas, or glioblastoma (GBM) [1]. Overall, ependymomas are the most common spinal glioma, comprising 50%–60%, followed by astrocytomas, comprising 20%–30%, with other histologic entities being far less frequent [2,3]. The intracranial:spinal ratio for astrocytomas and ependymomas is estimated to be 10:1 and 3:1-20:1 [4]. Although rare, spinal cord gliomas have significant associated morbidity and mortality. While spinal cord ependymomas and low-grade spinal cord astrocytomas (SCAs) have excellent rates of survival, overall survival (OS) for high-grade SCAs is poor secondary to their infiltrative nature and the inability to perform aggressive surgical resection (Fig. 1) [5]. One study of 106 evaluable SCA patients with median follow up time of 50.2 months after surgery, reported 5-year OS rates of 77%, 61%, and 7% for grades 2, 3, and 4 respectively [6]. Treatment options remain limited, focused on maximal safe surgical resection, radiation therapy and chemotherapy. As a result, additional therapies are urgently needed to prolong the lives of patients diagnosed with spinal cord gliomas, with immunotherapy providing a promising treatment option.
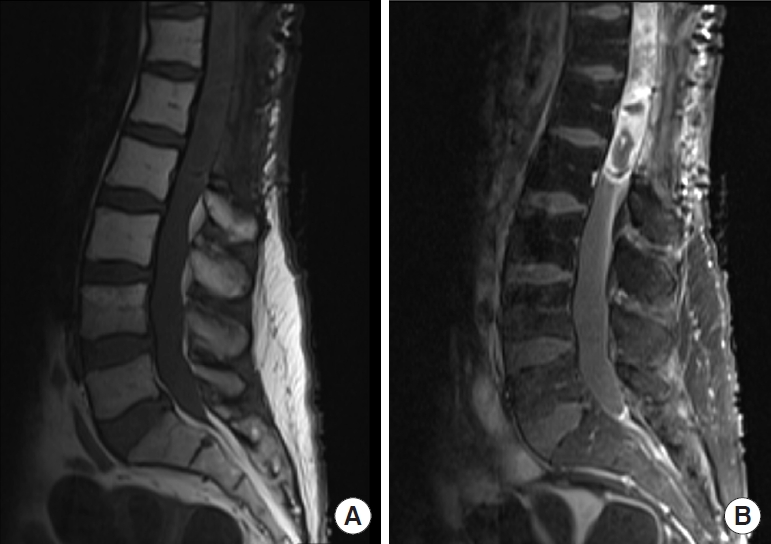
T1 with (A) and without (B) contrast demonstrating enhancing lesion of the conus medullaris with evidence of prior laminectomy. This patient had a subtotal resection of a World Health Organization grade III anaplastic astrocytoma 4 years prior and had undergone radiation and temozolomide therapy.
Alongside the advancements of surgical treatment, postoperative radiotherapy, and chemotherapy, immunotherapy has become an important fourth modality to treat malignancies. The goal of cancer immunotherapy is to generate a tumor specific immune response capable of selectively targeting and eliminating cancer cells. Although initially employed in the treatment of melanoma and hematologic malignancies, immunotherapy has been increasingly applied to gliomas following significant discoveries reversing the belief of an immunoprivileged CNS [7]. These novel advances in glioma immunotherapy include immune checkpoint inhibitors, chimeric antigen receptor (CAR) T therapy, and vaccine strategies (Fig. 2). Given the prevalence and dismal prognosis of intracranial GBM as compared to spinal gliomas, all of the landmark glioma immunotherapy trials are targeted to these patients. Use of immunotherapy in the treatment of spinal gliomas remains promising yet scarce due to considerable challenges. Below, we will summarize the current status of immunotherapy in intracranial gliomas, challenges when attempting to apply these to spinal cord gliomas, and ongoing solutions to these difficulties.
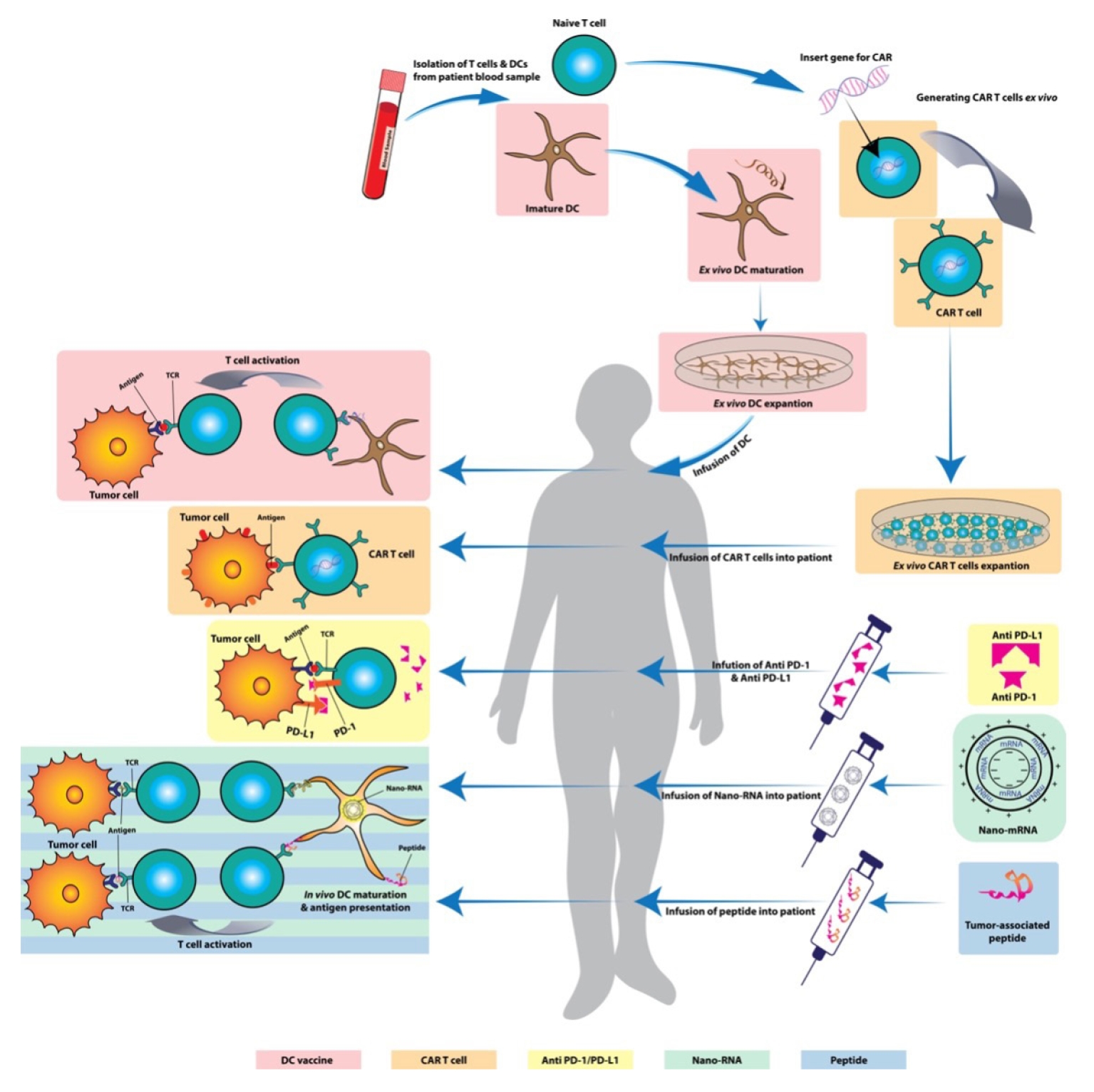
Summary of immunotherapy strategies. Cell-based therapies include dendritic cell (DC) and chimeric antigen receptor (CAR) T. These cells are isolated from a patient, modified and expanded ex vivo and infused back as systemic therapy. Drug-based therapies include antibodies to programmed death protein 1 (PD-1) and PD-1 ligand (PD-L1) which prevent cancer cells from evading immune recognition. Vaccine therapies include RNA and peptide-based strategies which trigger an in vivo immune response to the cancer antigens.
IMMUNE CHECKPOINT INHIBITORS: NIVOLUMAB AND PEMBROLIZUMAB
Immune checkpoints are negative-regulatory signaling mechanisms present within the normal immune system responsible for maintaining self-tolerance and preventing autoimmune diseases [8]. These mechanisms exist at the interface between T cells and normal host cells to alert T cells to self and subsequently attenuate the strength and duration of the response [8]. Many tumors, including gliomas, exploit the normally protective role of immune checkpoints to evade T-cell destruction. Among the inhibitory checkpoint ligands and receptors, programmed death protein 1 (PD-1) and PD-1 ligands (PD-L1 and PD-L2) are highly expressed within intracranial GBM [9,10]. In fact, PD-L1 is expressed by tumor cells, microglia, and peripheral-derived myeloid cells within the tumor microenvironment of approximately 61%–88% of intracranial GBM [11-13]. Therefore PD-1 and PD-L1/2 are potential targets for pharmacologic treatment of gliomas.
1. Anti-PD-1: Nivolumab
Murine glioma models treated with mouse monoclonal antibodies against PD-1, PD-L1, and CTLA-4 produced long-term tumor free survival in 50%, 20%, and 15% of treated animals, respectively [14]. Given the promising preclinical results of PD-1 inhibition in murine models, CheckMate 143 was initiated to determine whether nivolumab, a human PD-1 receptor inhibitor, improves survival in patients with recurrent GBM compared with bevacizumab, a VEGF inhibitor [15]. In this open-label, randomized, phase III clinical trial, 369 patients with GBM at first recurrence following standard radiation and temozolomide were randomized 1:1 to nivolumab 3 mg/kg or bevacizumab 10 mg/kg [15]. One hundred eighty-two and 165 received nivolumab and bevacizumab, respectively, every 2 weeks until disease progression, unacceptable adverse effects, or death [15]. Median time from initial GBM diagnosis to recurrence of the GBM was 10.1 months in the nivolumab arm and 8.5 months in the bevacizumab arm [15]. Median time from last radiotherapy dose to first dose of study drug was 8.8 and 6.9 months the nivolumab and bevacizumab arms, respectively [15].
Median OS (mOS) from time of randomization was similar between groups: 9.8 months with nivolumab versus 10.0 months with bevacizumab [15]. Median progression free survival (PFS) was 1.5 months with nivolumab and 3.5 months with bevacizumab [15]. mOS of both nivolumab and bevacizumab in this trial was comparable to the mOS of 7.2–9.1 months in previous prospective phase II trials evaluating bevacizumab monotherapy of 10 mg/kg every 2 weeks [16,17]. However, although PFS of bevacizumab in CheckMate 143 was similar to the PFS with bevacizumab monotherapy in these other phase II trials (3.7–4.2 months), PFS of nivolumab was notably less [16,17].
Unfortunately, this trial did not meet a primary endpoint of improved OS with nivolumab versus bevacizumab [15]. However, concurrent treatment with corticosteroids may have contributed to these less than promising results. 40% of patients treated with nivolumab and 42% of patients treated with bevacizumab were concurrently treated with corticosteroids, including 14% in both arms taking > 4 mg/day [15]. A subgroup analysis was performed to quantify mOS only of patients treated with corticosteroids within each arm to. The mOS was reduced in both arms, but more so in the nivolumab+corticosteroid treatment group. The reduced efficacy of nivolumab in steroid dependent patients led the authors of CheckMate 143 to hypothesize immunotherapy may be more efficacious in patients who require little to no steroids during their immune checkpoint treatment [15]. Patients requiring corticosteroids for symptomatic cerebral edema may possess more rapidly progressive disease and would not have sufficient time to benefit from immunotherapy [15]. In addition, the direct effect of corticosteroids on T-cell function may diminish the strength of the immune system and ability to benefit from immunotherapy [18]. Although CheckMate 143 did not have the desired result that many had hoped, the median duration of response was 11.1 months and 5.3 months in nivolumab and bevacizumab respectively, suggesting that responses were more durable in the nivolumab arm [19].
Neo-Nivo is a single-arm phase II clinical trial which sought to determine the feasibility, safety, and immunobiological effects of PD-1 blockade via administration of neoadjuvant and adjuvant nivolumab in the treatment of newly diagnosed and recurrent GBMs [20]. Thirty patients were included in the trial, 27 of which were cases requiring salvage surgical resection and the remaining were newly diagnosed in need of primary resection [20]. Prior to administration of nivolumab or surgical resection, baseline tumor tissue was acquired via biopsy or previous resection. After collection of this tissue, patients received 3-mg/kg nivolumab [20]. Two weeks later, patients underwent surgical resection and tumor specimens were collected to compare to pretreatment samples to detect the immunobiological effects of neoadjuvant nivolumab [20]. Patients received postsurgical doses of 3 mg/kg every 2 weeks until progression or unacceptable toxicity [20].
Treatment with nivolumab was well tolerated with only 27% of patients experiencing any grade immune related adverse events [20]. Of patients with recurrent GBMs, median PFS was 3.5 months, and mOS was 6.9 months [20]. Of the 3 cases enrolled with treatment-naive disease, 2 remained alive for over 2 years. No patients were excluded from the trial for treatment of cerebral edema with corticosteroids, no matter the dose, and 80% of patients in the trial required steroid treatment [20].
Much like CheckMate 143, little clinical benefit was observed for treatment of recurrent GBM in the Neo-Nivo trial [20]. However, multiple molecular analyses of pre- and posttreatment tumor samples indicated enhanced expression of proimmunotherapeutic molecules in the tumor microenvironment after treatment with neoadjuvant nivolumab [20]. Combinations with other immune and nonimmune agents may provide synergistic effects with neoadjuvant nivolumab required to demonstrate clinical efficacy [20,21].
2. Anti-PD-1: Pembrolizumab
In the Ivy Consortium Trial, pembrolizumab, a second PD-1 inhibitor, demonstrated promising results for the treatment of recurrent GBM. A total of 35 patients were enrolled in this multiinstitution, randomized, open-label pilot study. Sixteen patients were randomized into a neoadjuvant pembrolizumab group and 19 into an adjuvant only group. Two weeks prior to surgery, patients in the neoadjuvant group received 200 mg pembrolizumab. Tumor resection was then performed in participants. After recovery from surgery, all patients received pembrolizumab every 3 weeks until tumor progression or an adverse event requiring discontinuation [22]. Similar to the Neo-Nivo trial, tumor samples were collected pre- and posttreatment to evaluate molecular alterations in the tumor microenvironment.
Patients in the adjuvant only group had a mOS of 7.5 months, whereas those in the neoadjuvant group had a mOS of 13.7 months. Median PFS was 2.4 months in the adjuvant only group and 3.3 months in the neoadjuvant group; although, the authors recognized establishment of PFS may have been complicated by radiographic pseudoprogression. These substantial gains in mOS may be explained by the observed induction of antitumor changes within the tumor microenvironment. Neoadjuvant PD-1 blockade induced distinct tumoral gene expression changes relating to interferon and T-cell pathway induction and repression of the cell-cycle related transcriptional activity of tumor cells. Neoadjuvant tumor samples were also associated with focal upregulation of PD-L1 and CD8+ T-cell infiltration, and increased tumor-infiltrating lymphocytes (TILs) density was associated with increased OS.
Unlike patients enrolled in the CheckMate 143 and Neo-Nivo trials, patients in the Ivy Consortium Trial receiving high-dose dexamethasone (> 4 mg/day) were excluded in the final analysis to account for the potential immunosuppressive effects of corticosteroids [15,20,22]. This divergence in exclusion criteria may have contributed to the promising mOS in this cohort of patients. Corticosteroid use and appropriate exclusion criteria should be considered when treatment with immune checkpoint inhibitors is translated to spinal gliomas.
ADOPTIVE T-CELL TRANSFER: TIL, TCR T-CELLS, AND CAR T-CELLS
Other T-cell-centric therapies have evolved alongside immune checkpoint inhibitors with extremely promising results. Rather than using antibodies to directly stimulate in vivo mechanisms of T-cell regulation, adoptive T-cell transfer procures autologous T-cells, manipulates them ex vivo, and infuses them to ultimately function as “living drugs” to create long-term therapy [23]. In adoptive T-cell transfer, lymphocytes can be derived from naturally occurring T cells isolated from the resected tumor acquired from peripheral blood and genetically modified to be redirected to tumor antigens [23].
1. Tumor Infiltrating Lymphocytes
TILs are a heterogeneous cell population within the tumor primarily composed of T cells [23]. A portion of TILs express T-cell receptors (TCRs) on their surface which recognize tumorassociated antigens and become cytotoxic against malignant cells [23]. When isolated, these TILs can be expanded ex vivo and returned to the patient for T-cell directed tumor attack [23]. Although quite useful in melanomas, TILs have not shown to be of benefit in gliomas. Unlike melanomas, gliomas do not give rise to high numbers of infiltrating antitumor T cells making it difficult to isolate TILs for expansion [24]. Furthermore, not all infiltrating T cells are specific for tumor antigens and may simply be bystanders unable to mount a directed attack [25]. Only a few attempts have been made to treat patients with primary or metastatic brain tumors using TILs [26,27]. In one small prospective pilot study, six patients were infused with TILs acquired and expanded from resected, recurrent GBMs [26]. Although TILs were observed to be safe, no survival benefit was exhibited [26].
2. TCR T-Cell Therapy
While isolating TILs may be difficult, tumor-associated antigens are much more abundant and therefore more feasible to identify [23]. T cells in the peripheral blood can be genetically modified to redirect their specificity to these antigens [23]. Two approaches for genetic modification have been used: TCR engineered T cells and CAR T cells (CAR-T) [23]. TCR T cells are T cells bound with TCRs consisting of variable α and β chains specific to a tumor associated antigen [23]. TCR T cells recognize processed intracellular antigens presented by the major histocompatibility complexes (MHCs) on the surface of tumor cells [23]. Dependence on MHC warrants this approach less useful for gliomas that downregulate and decrease expression of MHC as a mechanism of immunoevasion [28]. Nonetheless, the desire to develop TCR T cells still remains because they have the unique advantage of identifying intracellular tumor antigens, expanding on the diversity of potential targets [29]. However, application of tumor-associated antigen-targeting TCR T-cell trials in various malignancies have been fraught with off-tumor, on-target toxicity [30-33]. There have been no clinical trials for TCR T-cell based therapies in gliomas, underscoring the need to identify neoantigen targets to pioneer and safely apply this therapy.
3. CAR T-Cell Therapy
CAR T-cell therapy was developed to overcome the challenge of MHC downregulation. CAR T-cells are autologous peripheral T-cells with addition of a CAR. A CAR is an extracellular, antigen binding domain, specific to tumor antigens or tumor associated antigens, translated in tandem with assorted intracellular signaling regions that when activated have differing effects on T-cell proliferation, function, and survival [34]. Because the properties of a CAR and TCR differ significantly, CAR T-cells can recognize surface antigens, including proteins and carbohydrates, without the need for processing and presentation with MHC [34]. CAR-T efficacy in intracranial gliomas has been demonstrated in preclinical trials targeting the following tumor-specific antigens: epidermal growth factor receptor variant III (EGFRvIII), erythropoietin-producing hepatocellular carcinoma A2 (EphA2), human epidermal growth factor receptor 2 (HER2), and interleukin receptor 13Rα2 (IL13Rα2) [35-38]. The positive results from these preclinical studies have led to a number of subsequent clinical trials. Many of these clinical trials are ongoing, but the handful of published data is extremely promising.
EGFRvIII is a constitutively activated, mutated form of the wild-type receptor located on 50% of gliomas [39]. This receptor is an ideal epitope for recognition by CAR T-cells because it is entirely absent from normal tissues and therefore extremely tumor specific with minimal toxicity to healthy cells. In a phase I trial, a series of 10 patients with recurrent, EGFRvIII-positive GBMs were treated with a single dose of intravenous EGFRvIII CAR T cells [40]. Seven out of 10 patients underwent posttreatment surgical intervention providing a window of opportunity to evaluate the microenvironment [40]. CAR T cells were present within the excised tumors along with a significant increase in new, immigrant, unmodified T cells [40]. Levels of EGFRvIII expression decreased posttreatment confirming successful CAR-T infiltration and activity [40]. Safety was the primary endpoint of this study and was demonstrated with no evidence of off-tumor toxicity or cytokine release syndrome [40]. The median time from diagnosis and initial resection to infusion was 358 days, with all patients receiving surgery, chemoradiation, and one or more cycles of adjuvant temozolomide as first-line standard treatment prior to recurrence and enrollment in this study [40]. Several patients received other lines of therapy prior to infusion, including bevacizumab, CCNU (lomustine), carboplatin, and dendritic cell (DC) vaccine therapy [40]. The mOS after infusion was 8 months in these 10 patients [40]. PFS was not evaluable because of the confounding factor of neurosurgical intervention in most of the subjects [40].
HER2, another receptor within the epidermal growth factor family, is expressed in 82.5% of intracranial GBMs making HER2 a more universal target than EGFRvIII [41]. However, its universality extends beyond GBMs and is also present in healthy epithelial cells, which carries a risk of off-target toxicity not exhibited by EGFRvIII [34]. After altering the exo- and endo-domains to reduce this risk, a phase I dose escalation study infused HER2 CAR T cells into 17 patients with HER2 positive, recurrent GBMs [42]. No dose limiting toxicities were observed, and mOS was 11.1 months after the first infusion and 24.5 months after diagnosis [42].
IL13Rα2 is a germline cytokine receptor found in 44%–47% of GBMs [43]. This receptor has shared expression in normal gametogenic cells within the testes but is not expressed at significant levels to cause off-target toxicity [43]. An IL13Rα2-specific CAR was developed with the ability to recognize IL13Rα2 and initiate cytolytic killing of GBM cells for a single-institution first-in-human pilot study [44]. Three patients were enrolled following initial diagnosis of high grade glioma [44]. At the time of first recurrence, the participant underwent resection of tumor along with placement of a Rickham reservoir and catheter [44]. Following recovery from surgery, the IL13Rα2-specific CAR T-cells were administered directly into the resection cavity via the indwelling catheter [44]. Escalating doses (5 × 107 –108) were administered intracranially over a 5-week period with a target total of 12 CAR T-cell doses [44]. Two out of the 3 patients received 12 doses, and the third received 11 [44]. Although a survival benefit could not be established with such a small cohort of patients, the 3 patients treated had a mean survival of 11 months after relapse and CAR-T treatment, with best response of 13.9 months [44]. In 2 of the 3 subjects, tumor recurrence occurred distant from the border of the resection cavity near T-cell infusion sites [44]. In the 1 patient with recurrence of the tumor adjacent to the treatment cavity, another craniotomy was performed 14 weeks after T-cell therapy and examination of this specimen showed significantly lower expression of IL13Rα2, implying selective targeting of IL13Rα2 – expressing tumor cells [44].
EphaA2 is a receptor overexpressed in GBMs promoting its malignant phenotype [36]. It is not expressed in normal brain tissue [36]. No clinical trial investigating CAR-T specific to EphA2 in gliomas has been completed to date, but there is one trial currently recruiting (NCT03423992).
VACCINES: TARGET ANTIGENS, VACCINE PLATFORMS, AND VACCINE VEHICLES
In 1953, observations in radiology highlighted a phenomenon known as the abscopal effect, where after radiation, there would be systemic regression of tumors and metastases in the nonradiated areas outside of the primary radiation field [45,46]. It was hypothesized that the radiation induces the apoptosis of tumor cells and release of tumor antigens which then prime the immune system for a systemic antitumor response [46]. This observation inspired the possibility of stimulating the immune system using exogenously introduced antigens and is the basis for the generation of anticancer vaccines. The abysmal survival rate of GBM has been largely attributed to the various mechanisms of GBM-induced immunosuppression, so clinical trials of vaccine-based immunotherapies have emerged as a potential treatment modality intended to overcome GBM pathogenesis and improve OS. Each vaccine trial can be categorized based on their target antigens, antigenic form, and vehicle of delivery. Target antigens are broadly classified as either tumor associated or tumor specific. Tumor associated antigens are antigens expressed in many cells throughout the body but are overexpressed in tumors [47,48]. In the case of GBM, these include survivin, Wilms tumor 1, and HER2. Conversely, tumor specific antigens include mutant proteins exclusively expressed by the tumor cells. Tumor specific antigens are considered ideal targets for vaccines because they are selectively expressed by gliomas cells and minimize off-target effects. Although challenging to identify in GBMs, several tumor specific antigens have been utilized, including EGFRvIII, H3.3K27M, CMV proteins, and isocitrate dehydrogenase (IDH) R132H [49]. The form which these antigens may be delivered also varies amongst vaccines. Each vaccine platform confers a slightly different mechanism of immunogenicity and mode of manufacturing also differs. There are various benefits and drawbacks associated with each platform and vehicle which must be considered.
1. Peptide Vaccines
Historically, live-attenuated or inactivated forms of microbial pathogens have been used for induction of antigen-specific responses that protect the host against subsequent infections. Based on the pathogen being used, vaccine formulation could contain anywhere between tens of to a few hundred proteins. However, immunity is usually dependent upon a mere epitope, and any additional proteins may induce allergenic or reactogenic responses. This created an interest in peptide vaccines containing only epitopes capable of inducing desirable T-cell and B-cell mediated responses. Peptides used in these vaccines are 10–30 amino acids designed to encompass a specific epitope of tumor specific antigens. GBMs are notorious for exhibiting a low mutational load compared to other solid tumors which makes identifying tumor-specific antigens challenging [50]. Nonetheless, several antigens have been identified as potential targets, and the peptide vaccines, rindopepimut, IMA950, IDH1, and H3.3K27M26-35 are currently being explored.
ACT IV was a randomized, placebo controlled, phase 3 clinical trial to assess whether the addition of rindopepimut, a peptide vaccine targeting EGFRvIII, to standard temozolomide chemotherapy increased OS compared with that for temozolomide alone in patients with newly diagnosed EGFRvIII-expressing gliomas [48]. 165 hospitals in 22 countries contributed 745 eligible patients who were randomized to receive rindopepimut (n=371) or keyhole limpet hemocyanin, serving as the control (n=374) [48]. The primary endpoint was OS defined as time from randomization to death in patients with newly diagnosed EGFRvIII-positive GBM [48].
Although rindopepimut elicited a strong humoral response, with treated patients reaching a median anti-EGFRvIII antibody titer of 1:25,600, it conferred no significant improvement in PFS or mOS compared to controls [48]. The mOS in the rindopepimut group was 20.1 months versus 20.0 months in the control group [48]. Median PFS was 8.0 months in the rindopepimut group and 7.4 months in the control group [48]. These unexpected results were disappointing to the community, given the multiple, independent, previously completed phase II studies suggesting survival advantage within the ACTIVATE, ACT II, and ACT III trials [51-53].
Of note, patients in the control arm fared substantially better than matched controls available at the time of the study design resulting in an insignificant difference in mOS between the 2 arms [48,51-53]. A plausible explanation as to why ACT IV did not agree with earlier studies include a skewed interpretation of the results of ACTIVATE, ACT II, and ACT III given these trials lacked randomization and comparison of survival outcomes were to potentially outdated historical controls [48,51-53]. If this is the case, rindopepimut would have appeared more efficacious in the initial trials, and the control group in ACT IV may have fared well simply due to optimization of standards of care and improvements in outcome over time. Although EGFRvIII peptide immunization did not demonstrate survival advantages, the monumental effort of the ACT IV trial aligns with the current direction of clinical efforts towards personalized immunotherapy for the treatment of patients with GBM.
Whereas rindopepimut targets a single antigen, several multiantigen vaccines are in early phase trials to the avoid potential loss of expression with a single antigen treatment. A phase I/Ib trial produced personalized vaccines for eight patients with newly diagnosed methylguanine methyltransferase (MGMT)-unmethylated GBM [54]. Surgically resected tissue and matched normal cells were analyzed using whole-exome sequencing to identify coding mutations as potential neoantigens [54]. Eight vaccines were produced containing a median of 12 identifiable antigenic peptides with a median amino acid length of 24 per peptide [54]. Prior to administration of vaccines, patients received conventional radiation therapy administered at 180–200 cGy per fraction daily for five days per week to a total of approximately 60 Gy. Temozolomide chemotherapy was not administered to any patients as all tumors lacked methylation of the MGMT promoter. Median time from surgery to first vaccination was 19.9 weeks. Median PFS and OS were 7.6 and 16.8 months, respectively. Of note, only patients who did not receive dexamethasone during vaccine priming generated a robust de novo immune response of circulating polyfunctional CD4+ and CD+ T cells against the neoantigens [54]. Systemic depletion of naive and memory CD4+ and CD8+ T cells associated with dexamethasone poses a significant challenge to immunotherapy, including vaccinations [55].
Because of the relatively small size of peptide vaccines, they are often weakly immunogenic by themselves and chemically unstable. Therefore, they commonly require carrier molecules to induce a robust immune response and remain stable. Peptide antigens may be encapsulated by nanoparticles (NPs) to increase the longevity of the antigen, and DCs may be loaded with peptides and administered to patients for activation of T cells.
2. RNA Vaccines
The use of nucleic acid vaccines, including RNA vaccines, have emerged as a promising alternative to conventional vaccine approaches, as demonstrated by the Pfizer-BioNTech and Moderna mRNA coronavirus disease 2019 vaccines. This platform combines the efficacy of in situ expression of antigens with the safety of inactivated/subunit vaccines. The desired mRNA can be injected, and the subsequent proteins are expressed in the cells of the patient [56,57]. The mRNA will encode the antigen of interest, 5’ and 3’ untranslated regions, a 5’ cap, and a poly A tail [56]. Translation occurs in the cytosol without the need to transport to a specific organelle and normal degradation will follow decreasing the chance of toxicity [56]. RNA vaccines are in the early stages of development as a potential treatment of GBM patients. “Naked” RNA vaccines are injected directly, formulated only in buffer without a carrier [58]. These remain limited by the short extracellular half-life of naked mRNA due to rapid degradation by ubiquitous RNases. For this reason, RNA vaccines rely heavily on vehicles for delivery [58]. RNA vaccine clinical trials against GBMs have included DCs and NPs as carriers.
DCs present antigens to CD8+ cytotoxic T cells, natural killer cells, natural killer T cells, and CD4+ helper T cells to stimulate an immune response [59]. DCs may be isolated, loaded an RNA sequence ex vivo, and injected into patients resulting in a rapidly induced antigen-specific response in vivo [59]. Potency of tumor-antigen-specific DC may be increased with additional preconditioning of the vaccine site with a potent recall antigen, such as tetanus-diphtheria toxoid (Td) [60]. Cytomegalovirus pp65 is a protein that is expressed in over 50%–70% of GBMs but not in healthy brain tissue providing a tumor-specific target which can be pulsed into DCs in the form of RNA [61]. A randomized and blinded phase I clinical trial enrolled 12 patients with newly diagnosed GBM to evaluate the influence of vaccine site preconditioning with Td on Cytomegalovirus pp65 RNA-pulsed DC migration. After completion of standard therapy, patients were randomized 1:1 to receive preconditioning with autologous, unpulsed DCs (serving as a control) or Td before vaccination with Cytomegalovirus pp65 RNA-pulsed DCs [60]. From time of diagnosis, median PFS and OS in the control preconditioned unpulsed DC cohort was 10.8 and 18.5 months, respectively. Conversely, from time of diagnosis, median PFS and OS in the Td preconditioned group was 31.3 and 34.9 months, respectively. Thus, the median PFS and OS for the DC cohort were consistent with patients treated with the standard of care, while patients preconditioned with Td far exceeded it [60]. Unfortunately, DC vaccines have been confronted with logistical challenges in development, manufacturing, and marketing [62]. Alternatively, commercially available and clinically translatable NPs can be combined with tumor-derived RNA.
3. Nanoparticle Vaccines
Commercially available and clinically translatable NPs can be combined with tumor-derived RNA to provide an avenue to overcome the challenges of manufacturing vaccines with DC carriers [62]. RNA can be extracted from tumor samples, amplified, and combined with nanoliposomes to create personalized RNA-NP vaccines [62]. These can be generated within several days and have excellent scale-up capacity [62]. Furthermore, RNA-NPs, have the benefit of bypassing MHC class restriction, targeted localization, and immunogenic potential of RNA as a toll-like receptor agonist [63]. Trials of RNA-NP in mice and canines with gliomas have been promising [62,63]. Although treatments have yet to performed on humans with gliomas, the first in human phase I/II study of RNA-lipid particle (RNA-LP) vaccines for newly diagnosed adult MGMT unmethylated GBM will begin recruiting October 2021 with an estimated study completion date of October 2022 (NCT04573140). The clinical trial will consist of 2 strata divided into adult GBM patients and pediatric high-grade glioma patients in the newly diagnosed setting. The adult stratum of the clinical trial will enroll a maximum of 28 patients who will undergo surgery and chemoradiation prior to vaccine administration. Following surgical resection, tumor material will be collected for RNA extraction, amplification, and liposomal loading for vaccine production. RNA will consist of autologous total tumor mRNA and pp65 full length lysosomal associated membrane protein mRNA. RNA-LP vaccination will begin within 4 weeks following radiation. Patients will receive three RNA-LP vaccines every 2 weeks before beginning 12 cycles of adjuvant monthly RNA-LP vaccines for a total of 15 vaccines.
CHALLENGES IN THE APPLICATION OF IMMUNOTHERAPY TO SPINAL CORD GLIOMAS
Although spinal cord gliomas represent a rare group of malignancies when compared to their cranial counterparts, their associated morbidity and mortality are significant. Spinal cord ependymomas have 5-year survival rates approaching 100%, and low-grade SCAs have an OS of 90% at 50 months due to excellent local control rates with gross total resection [64,65]. Conversely, OS at 5 years for high-grade SCAs approaches a mere 15%–28% [5]. The infiltrative nature of high-grade SCAs and eloquent structure of the cord limits the achievable extent of resection (EOR). Aggressive surgical removal, an important intervention for intracranial gliomas, is not associated with significant survival benefit and not routinely recommended in high-grade SCAs due to morbidity [5]. Following biopsy and/or resection, radiation therapy is considered the standard adjuvant treatment [66]. However, given the aggressive nature and dismal prognosis of high-grade SCAs, systemic treatment options must be considered [66]. Temozolomide and PCV (procarbazine, lomustine, and vincristine) have been used with some effectiveness in the recurrent setting, though additional treatment options remain scarce and limited to case reports [66]. GBMs are at the forefront of immunotherapy, as evidenced by the extensive body of literature on the numerous mechanisms mentioned above. Blueprints of various immunotherapies have been derived from the research conducted on GBMs and applied to spinal gliomas. As these immunotherapies have been explored in the context of SCAs, notable differences have emerged between intracranial and spinal gliomas. These divergences pose significant challenges in the application of immunotherapy to spinal cord gliomas including low incidence, scarcity of targetable antigens, delivery across the blood-spinal cord barrier, immunosuppressive nature of the spinal cord tumor microenvironment, and neurotoxic treatment effects. Each must be overcome with novel solutions.
1. Low Incidence
A significant challenge in developing therapies for these tumors is that primary spinal cord tumors are exceedingly uncommon, with an incidence of 0.22 spinal cord gliomas per 100,000 person-years [67]. Given this rarity, it is very difficult to organize large randomized controlled trials. Regional and international trials would be helpful to address the issue of incidence as the patient pool would significantly increase. However, there are obvious logistical challenges in organizing these large trials, so most clinical studies, albeit very few, are performed at single centers with a very limited number of cases. The bulk of research specific to spinal gliomas has been performed on animal models, many of which use rat glioma cell lines rather than xenotransplantation models due to difficulties with rejection in the spine [68]. As opposed to the robust conclusions to be drawn from the many randomized clinical trials for intracranial GBMs, the conclusions drawn from small spinal glioma trials or rat glioma models are often incomplete or not clinically applicable.
2. Paucity of Specific Spinal Glioma Antigens and Antigenic Escape
The paucity of identifiable tumor specific antigens in spinal gliomas, especially high-grade SCAs, severely restricts the development of immunotherapies. TCR T cells, CAR T cells, and vaccines dependent on successful identification of antigens. Spinal gliomas were originally assumed to have similar genetic mutations and targetable antigens as their intracranial counterparts [69]. This notion was challenged when minimal MGMT promoter methylation in spinal gliomas was found to be the reason adjuvant chemotherapy and radiation lacks benefit in the spine [70]. Recent advances in molecular studies have uncovered the genomic landscape is actually quite different in spinal gliomas [69]. Mutations in the IDH1 gene frequently occur in brain astrocytomas, but the incidence of IDH1 mutations in spinal astrocytomas has been found to be quite low, with several studies observing no IDH1 mutations within their cohorts of SCAs [69,71-74]. The H3.3K27M26-35 variant is one of the few mutations which has been implicated in the tumorigenesis of both intracranial and SCAs. Although the general concepts of intracranial immunotherapy may be applicable to gliomas in the spine, alternative antigens must first be identified.
A contributing factor to the lack of identifiable antigens is the minimal tissue to be acquired from spinal gliomas to study and derive antigens from. The area of tumor volume able to undergo resection is far less in spinal gliomas when compared to intracranial masses. A single institution retrospective review analyzing EOR examined median tumor volumes of 115 patients with intracranial GBMs at the time of presentation was 36.5 cm3 as opposed to intramedullary spinal lesions with a mean volume of 0.55 cm3 [75,76]. For high-grade SCAs, extensive gross total resection is contraindicated because the risk of surgically acquired neurological deficits is far greater than any associated survival benefit [77]. The infiltrative nature of high grade spinal gliomas makes discerning the borders of the lesion difficult, further reducing the possibility of sufficient tumor harvest [77]. The combination of small volume and infiltrative borders provides little tissue to work with to identify antigens. Advancements in other immunotherapeutic treatments which require generous amounts of tumor, such as harvesting TILs or acquiring tumor lysate for vaccines, are slow to progress due to lack of tumor volume. Additionally, there may simply be less genetic changes overall in spinal tumors due to the small spinal canal volume and early detection of tumors due to symptoms when compared to intracranial lesions [78].
With the limited knowledge of identifiable spinal glioma antigens, scientists are often pressed to use single antigen strategies within immunotherapy [79,80]. As opposed to DC and peptide vaccines generated with multiple antigens, single antigen therapies increase the probability of antigen escape, an important cause of drug resistance and tumor relapse in gliomas. Our predicament of few available antigens may be further exacerbated when these targets become obsolete due to resistance. A sense of urgency must be adopted to identify new antigens specific to spinal gliomas at a rate outpacing the opposing antigenic escape.
3. Blood Spinal Cord Barrier
For a clinical benefit to be observed, immunotherapies must overcome the impermeability of the blood-spinal cord barrier (BSCB). The BSCB is the functional equivalent of the blood-brain barrier (BBB) as it fosters a regulatory and protective microenvironment for cellular constituents of the spinal cord. The BSCB shares the same non fenestrated endothelial cells and accessory structures as the BBB, so the BSCB was assumed to be a morphological extension of the BBB into the spinal cord. However, new data suggests unique morphological differences in these endothelial cells that create a relatively independent physiologic entity. Several studies have indicated increased permeability of the BSCB, as compared to the BBB, partially explained by differences in cell junction protein expression of endothelial cells [81-83]. This variation may prove to be a beneficial difference, but clinical trials with effective administration of immunotherapy to intracranial GBMs cannot be assumed to be translatable to the spine. Alternative routes of administration have been investigated to improve locality and specifically target spinal lesions. These include intrathecal, intraventricular, and intranasal administration [84-87]. Intraventricular administration is an option for cases with both intracranial and spinal cord lesions [86]. An intraventricular catheter may be placed at the time of intracranial resection [86]. Intranasal administration is a novel, noninvasive method of administration that may target drugs to the brain and spinal cord via the olfactory and trigeminal nerves [84,85]. Drug delivery systems have also been investigated as a means to transport treatments with otherwise poor permeability across the BSCB. NPs represent one of the most promising systems capable of this task [88].
4. Immunosuppressive Nature of Tumor Microenvironment
Not only does the tumor microenvironment play a crucial role during tumorigenesis, growth, and metastasis, it has profound effects on therapeutic efficacy. Major constituents of the tumor microenvironment include vasculature, immune cells, tumor-associated endothelial cells (TECs), cancer-associated fibroblasts, and an abnormal extracellular matrix [89]. The tumor microenvironment differs between the brain and spinal cord. Glioma cells were isolated from brain and cord tumors then transplanted in naive rat spinal cords and brains. The resulting tumors’ phenotypes were consistently predicted by the tissue into which they were transplanted rather than by the tissue of origin [78]. Tumors induced in the spinal cord were less likely to have necrosis and had differential platelet derived growth factor expression. Decreased necrosis and hypoxia within spinal cord gliomas seems like an advantage. However, this finding does not translate into increased sensitivity to standard therapies.
TECs receive signals from nearby tumor cells to trigger an immunosuppressive phenotype, as opposed to the immunosupportive nature of normal endothelial cells. TECs will express PD-L1 thereby increasing interactions with PD-1 and inducing the immune checkpoint [90]. T-cell function is suppressed, and the immune response is dampened to favor tumor growth [90]. Twenty percent of spinal gliomas express PD-L1, possibly through glioma TECs [91]. Although immune checkpoint inhibitors may be increasingly useful in this subset of spinal gliomas, other immunotherapies may be less effective or require adjuvant checkpoint therapy.
5. Neurotoxic Treatment Effects
The clinical benefits derived from immunotherapy are often dependent on inflammatory mechanisms of action. This presents unique challenges for spinal gliomas within a very narrow spinal canal. In a study with anti-GD2 CAR T-cell therapy for H3K27M+ diffuse midline gliomas, orthotopic mouse xenograft models for the thalamus, spine, and pons were developed [79]. Treatment of the thalamic xenograft model resulted in substantial treatment related neurotoxicity during the period of maximal therapeutic effect [79]. The robust neuroinflammatory response caused significant edema in a neuroanatomical location intolerant of swelling [79]. The spinal cord model was generated by injection of the graft into the medulla of the model to avoid paralysis induced by injection into the cord [79]. Neurotoxicity was not observed in the spine model, possibly due to the alternative location of the graft [79]. Although this study represents a promising advancement in the application of immunotherapy to spinal gliomas, safety cannot be assumed. If these treatments are translated clinically, very close clinical monitoring will be necessary to avoid permanent neurologic deficits.
OVERCOMING CHALLENGES WITH NOVEL SOLUTIONS
1. Overcoming the BSCB
Locoregional, including intrathecal and intraventricular, routes of administering CAR T cells have demonstrated efficacy against medulloblastomas and posterior fossa ependymomas [87]. Seven hundred sixty-three human primary medulloblastoma samples and 100 human primary ependymoma samples, 15 of which were derived from the spinal cord, were analyzed for CAR T targets [87]. EPHA2, HER2, IL13Rα2, and combination (TRI) CAR T cells were synthesized due to elevated expression of these targets in both tumor types [87]. Patient derived medulloblastoma and ependymoma cell lines were xenografted into the cerebellum of mice [87]. To identify the optimal approach for delivery of the CAR T cells, a single dose IV tail infusion was compared with left ventricular and intrathecal infusions [87]. There was a significant increase in survival of models receiving locoregional therapy vs intravenous administration [87].
NPs represent one of the most promising drug delivery systems capable of crossing the BSCB due to their favorable composition. Novel multifunctional liposomes with a hydrophobic core for drug encapsulation and a hydrophilic coat conjugated with biological materials, polyethylene glycol (PEG) and transactivating-transduction (TAT), were injected through the caudal vein of mice. PEG reduces recognition by mononuclear phagocytes, and TAT induces an endocytic event at the plasma membrane. Liposomes with these modifications demonstrated excellent uptake across the BSCB and exhibited prolonged circulation time. Induction of antitumor immunity using relevant RNA-loaded nanoliposomes has been achieved in an intracranial GBM mouse model and will be applied to humans in a phase I/II clinical trial enrolling soon (NCT04573140), but no attempts have been made to create a vaccine incorporating TEG-TAT multifunctional lysosomes with antigen specific RNA to spinal gliomas [62].
2. Molecular Markers of Spinal Gliomas
Where discovery of molecular markers was historically dependent on histological analysis, genetic and genomic studies allow for increased capabilities of identifying tumor markers even with minimal tissue [73]. Although scarce, promising antigens have been identified within spinal glioma tissue with genetic and genomic studies. H3.3K27M is a truly glioma specific antigen with overlap in expression between intracranial and spinal gliomas [69,88,92,93]. This antigen was discovered during the pursuit to understand the molecular pathogenesis underlying pediatric gliomas [94]. Missense mutations in the H3F3 gene, or less commonly in the related HIST1H3B gene, encode the histone 3 variant H3.3 and lead to amino acid substitutions at 2 critical positions within the histone tail: lysine at position 27 for methionine (K27M) or glycine at position 34 for arginine or valine (G34R/G34V) [94]. These substitutions invoke disruption of oncogenes and tumor suppressor genes and may play a role in gliomagenesis [95]. The K27M mutation most commonly occurs in pediatric gliomas with infiltrative, predominantly astrocytic differentiation, in midline locations (thalamus, brainstem, and spinal cord) [96]. In 2016, the World Health Organization termed this newly defined subset of tumors diffuse midline gliomas, H3 K27M-mutants, which included tumors previously referred to as diffuse intrinsic pontine gliomas [97]. Historically H3K27M-mutant DMGs have been diagnosed primarily in children, although they can be present in adults [98].
Because histone genes are highly conserved throughout eukaryotes, and no other human disorders have been associated with mutations in the H3.3 histone, this mutation was particularly intriguing as a potential immunotherapeutic target [94]. TCRs recognizing H3.3K27M26-35 have demonstrated efficient targeting and cytotoxic behavior in preclinical, in vivo models [99]. Chheda et al. [99] created DMG mouse models via intracranial injection of U87H3.3K27M cells. After tumor establishment, xenografted intracranial glioma mice models were infused with H3.3K27M26-35 TCR T cells, and bioluminescence imaging indicated significant reduction in the tumor burden of these mice when compared to controls. The impact of therapy on long-term survival was unable to be evaluated due to onset of graft-versus-host disease after day 31 in mice receiving TCR-transduced T cells [99]. Although these DMG mice models were xenografted intracranially, the promising H3.3K27M-specific T-cell response confirmed the immunogenicity of this epitope and promoted future applications [99].
H3.3K27M is a promising antigen for utilization of immunotherapy in the spine because 40%–50% of diffuse spinal cord astrocytic tumors exhibit the mutation, and H3.3K27M has also been detected in other spinal gliomas, such as a spinal pilocytic astrocytoma [69,100,101]. Two other antigens have been identified as potential targets: the telomerase reverse transcriptase gene promoter (TERTp) mutation and the TP53 mutation, found in 22.4% and 50% of spinal cord gliomas, respectively [69,74].
APPLICATIONS OF IMMUNOTHERAPY IN THE SPINE: CASE REPORTS AND CLINICAL TRIALS
Identification of novel targets, innovative routes of immunotherapeutic administration, and the success of numerous immunotherapies in intracranial gliomas have led to the application of immunotherapies for the treatment of intramedullary lesions of the spine in humans. Although many are metastatic and nonglioma in nature, the successful use of checkpoint inhibitors, vaccines, and CAR T-cell therapy for lesions of the spine, regardless of lesion classification, supports the notions that the BSCB can be breached, the spinal cord microenvironment is not entirely immunosuppressive, and the neuroinflammatory response associated with treatment is tolerable even in this eloquent location. Of the few clinical trials and case reports specifically on the treatment of spinal gliomas with immunotherapy, results are extremely promising and support continued efforts.
1. Regression of a Non-Small-Cell Lung Cancer Spinal Cord Metastasis With Nivolumab
Lung cancer is the most common primary tumor associated with intramedullary spinal cord metastasis (ISCM) accounting for over 50% of cases [102]. When it occurs, it is associated with very poor prognosis and is traditionally treated with corticosteroids and radiation [102]. Although nivolumab has been approved for the treatment of non-small cell lung cancer (NSCLC) and data on its efficacy against brain metastases are encouraging, its efficacy against parenchymal spinal cord metastasis is unknown. Phillips et al. [103] reported a patient with advanced-stage NSCLC and ISCM treated with nivolumab and exhibited subsequent regression of her ISCM. A 69-year-old woman with a 50-pack-year tobacco history presented with a left upper lobe mass, mediastinal adenopathy, hepatic adrenal, and splenic metastases, and extensive skeletal metastatic disease [103]. A screening brain magnetic resonance imaging (MRI) one month later demonstrated 35 sub centimeter intracranial nodules consistent with metastases [103]. CT guided lung biopsy of the mass confirmed NSCLC [103]. She underwent CyberKnife radiotherapy to less than half of the intracranial lesions and received 5 cycles of carboplatin/pemetrexed followed by maintenance pemetrexed [103]. There was initially interval decrease of the treated intracranial lesions, but subsequent imaging 6 months after radiotherapy demonstrated marginal growth of brain metastases [103]. Nine months after diagnosis, nivolumab 3 mg/kg every 2 weeks was initiated [103]. A brain MRI 2 weeks after treatment continued to show slight increase of the previously identified intracranial metastases as well as an incidentally discovered 3-mm cervical intramedullary signal at the bottom of the sagittal brain sequence. A total spine MRI confirmed a 4-mm intramedullary metastasis at C3–4 with extension of edema [103]. The patient remained asymptomatic and continued with nivolumab. Two months after the initiation of nivolumab, interval imaging demonstrated unchanged size of the cord metastasis with significant increase in severe cord edema extending throughout the cervical cord. Nonetheless, she remained clinically stable in the absence of corticosteroids. Following her tenth dose of nivolumab, a brain MRI demonstrated decrease of the majority of her brain lesions with significant improvement of vasogenic edema [103]. A cervical spine MRI demonstrated decrease size in the C3–4 lesion with decrease in cord swelling and edema. After 7 months of treatment with nivolumab, a cervical spine MRI showed complete resolution of the spinal metastasis, which persisted for the duration of surveillance for an additional 6 months [103].
2. Nivolumab as Treatment for a Spinal Melanocytoma
A second case report demonstrated success of nivolumab for the treatment of a spinal melanocytoma. A 70-year-old man presented with motor sensory deficit in his left lower extremity and bilateral upper extremity neuralgia. A spine MRI demonstrated an intramedullary tumor at C7–T1. In the initial work up, there was no evidence of metastatic spread of a primary lesion. The tumor was excised and deemed to be a meningeal melanocytic tumor based on the microscopical examination, immunohistochemistry, and molecular studies. He received ten sessions of radiotherapy and 6 cycles of nivolumab at a dose of 3 mg/kg. He exhibited stability of his disease for 13 months following treatment with nivolumab before follow-up MRIs revealed enlargement of the residual tumor. He underwent a second excision and 6 cycles of temozolomide with tumor recurrence and eventual death after becoming septic and developing a pulmonary embolism. Although the patient died because of complications secondary to tumor evolution, the interval stability of his disease with immunotherapeutic treatment was a promising development in the treatment of spinal cord tumors.
3. H3.3K27M-Specific Vaccine Responses in Diffuse Midline Gliomas
After the discovery of the H3.3K27M antigen, it was quickly utilized as a target for TCR T-cell therapy, CAR T-cell therapy, and incorporated into peptide vaccines with little off-target toxicity and promising success in diffuse midline gliomas, predominantly intracranially but in the spine, as well [29,79,99,104]. In Chheda et al. [99], although the diffuse midline glioma mice models were xenografted intracranially, the promising H3.3K27M-specific T-cell response confirmed the immunogenicity of this epitope. This subsequently led to a multicenter pilot study to evaluate the safety, immunoreactivity, and preliminary efficacy of a vaccine using a 10 amino acid peptide spanning position 26-35 of the H3.3K27M protein (H3.3K27M26-35) in diffuse midline gliomas [104]. In the small pilot trial, H3.3K27M,26-35 along with helper tetanus toxoid and poly-ICLC, was administered to 29 patients with diffuse midline gliomas [104]. Patients were stratified based on location of their tumors and outcomes were reported per stratum. Nineteen patients with tumors located in the pons were enrolled in stratum A, and 10 patients were enrolled in stratum B [104]. DMGs in stratum B included 7 thalamic tumors, 2 pontine-centered tumors with extension into the cerebellum and cervical spine, and 1 spinal cord tumor in the thoracic spine [104]. Outcomes were not reported for the individual patient with the spinal cord tumor but rather for the entire stratum [104]. Nonetheless, the treatment was well tolerated and exhibited promising clinical efficacy for patients in stratum B. No grade 4 treatment-related adverse events were reported [104]. The rate of OS at 12 months for patients in stratum B was 39%, and the median PFS was 3.5 months for stratum B [104]. Seven of the 29 patients were deemed “responders” if they exhibited a 25% postvaccination increase in H3.3K27M26-35-reactive CD8+ T cells [104]. These seven patients exhibited a median OS of 16.3 months post injection as opposed to the OS of 9.9 months in the “nonresponder group” [104]. These results have led to a follow-up multicenter phase I/II study evaluating the safety, immunoreactivity, and efficacy of the H3.3K27M26-35 peptide vaccine given in combination with nivolumab to patients with newly diagnosed diffuse midline gliomas, including spinal tumors (NCT02960230).
4. Regression of Spinal GBM Metastases After CAR T-Cell Therapy
A case of a 50-year-old man presented with a newly diagnosed right temporal lobe GBM [86]. He received standard of care therapy consisting of resection, radiotherapy, and temozolomide, but six months after diagnosis, his MRI demonstrated evidence of recurrence with five progressing intracranial tumors [86]. The patient was enrolled in a phase I clinical trial for patients with recurrent gliomas to receive IL13Rα2 targeted CAR T cells (NCT02208262). Three of the 5 intracranial tumors were resected, and a Rickham device was implanted into the resected cavity of the largest tumor [86]. The patient received weekly intracavitary infusions of IL13Rα2 targeted CAR T cells via the catheter for 6 cycles, and there was no evidence of disease progression from this site for more than 45 days [86]. However, an interval MRI demonstrated progression of the two nonresected tumors and 2 new tumors near the resected lesions [86]. Additionally, there were several new metastatic lesions in the spine, including one tumor 18 mm and several smaller tumors ≤ 4 mm [86]. These results suggested that although administration of CAR T cells may have prevented local recurrence at the site of the intracavitary infusion, the infusions were not sufficient to effectively control progression at distant locations [86]. Based on the rationale that delivery of the immunotherapy into the cerebrospinal fluid would improve trafficking to these new sites of disease, a second catheter was placed in the lateral ventricle [86]. Ten intraventricular infusions were performed without any other therapeutic interventions [86]. There was a substantial reduction in the size of all intracranial and spinal lesions after the first three infusions [86]. After the fifth, all tumors had decreased by 100% [86]. All lesions continued to resolve throughout the remaining five infusion infusions [86]. No tumors, including the metastatic spinal lesions, were measurable by MRI or detectable by positron emission tomography [86]. During intraventricular treatments, dexamethasone was tapered to elimination, and the patient returned to work [86]. This response was sustained for 7.5 months before the patient experienced a recurrence at 4 distinct, nonadjacent sites, both intracranially and spinally [86]. The cause of recurrence is under investigation, but early results suggest reduced expression of IL13Rα2 within the new lesions [86]. The intraventricular route of administration, involvement of spinal lesions, and remarkable clinical response in this patient makes IL13Rα2 targeted CAR T cells especially promising for the application to spinal gliomas.
CONCLUSION
High-grade spinal cord gliomas are aggressive lesions with malignant behavior. Due to their invasive growth and low incidence when compared to intracranial gliomas, the current standard of care does not significantly increase survival and improvements upon this have been slow. Immunotherapy has been incredibly promising for other cancers and has been increasingly applied to intracranial gliomas with promising results. Application of these therapies have rarely been applied to spinal gliomas due to the challenges of penetrating the BSCB, antigenic escape, lack of targetable antigens, the unique tumor microenvironment, and spinal cord neurotoxicity. Nevertheless, unique antigens have been discovered, novel administration techniques and drug delivery platforms have been explored to overcome the BSCB and beneficial differences in spinal gliomas have been postulated. Together, these imply appropriate application of immunotherapy to spinal gliomas may be the next meaningful therapy in the treatment of spinal gliomas.
Notes
Conflict of Interest
The authors have nothing to disclose.
Funding/Support
This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Author Contribution
Conceptualization: KM, KP, DH, MR, AG; Data curation: C Grady, KM, KP, MR, AG; Formal analysis: C Grady, KM, AG; Methodology: KM, MR, AG; Project administration: DH, MR, AG; Visualization: FD; Writing - original draft: C Grady, KM, AG; Writing - review & editing: C Grady, KM, KP, DH, MR, AG.