Pathological Classification of the Intramedullary Spinal Cord Tumors According to 2021 World Health Organization Classification of Central Nervous System Tumors, a Single-Institute Experience
Article information

Abstract
According to the new 2021 World Health Organization (WHO) classification of tumors of the central nervous system (CNS) the classification of the primary intramedullary spinal cord tumors (IM-SCT) follows that of CNS tumors. However, since the genetics and methylation profile of ependymal tumors depend on the location of the tumor, the ‘spinal (SP)’ should be added for the ependymoma (EPN) and subependymoma (SubEPN). For an evidence-based review, the authors reviewed SCTs in the archives of the Seoul National University Hospital over the past decade. The frequent pathologies of primary IM-SCT were SP-EPN (45.1%), hemangioblastoma (20.0%), astrocytic tumors (17.4%, including pilocytic astrocytoma [4.6%] and diffuse midline glioma, H3 K27-altered [4.0%]), myxopapillary EPN (11.0%), and SP-subEPN (3.0%) in decreasing order. IDH-mutant astrocytomas, oligodendrogliomas, glioneuronal tumors, embryonal tumors, and germ cell tumors can occur but are extremely rare in the spinal cord. Genetic studies should support for the primary IM-SCT classification. In the 2021 WHO classifications, extramedullary SCT did not change significantly but contained several new genetically defined types of mesenchymal tumors. This article focused on primary IM-SCT for tumor frequency, age, sex difference, pathological features, and genetic abnormalities, based on a single-institute experience.
INTRODUCTION
The spinal cord belongs to the central nervous system (CNS) and is a tubular structure that leads to the medulla oblongata, from which any tumor arising from the brain can develop. All intradural extramedullary spinal tumors (EM-SPT) and intramedullary spinal cord tumors (IM-SCT) are rare and account for 2%–4% of CNS tumors [1]. The incidence of spinal cord tumors in Seoul National University Hospital (SNUH) is similar to that reported previously [2]. Reported IM-SCT accounts for approximately about 10% of spinal cord tumors [1]. For the classification of the IM-SCT according to the updated World Health Organization (WHO) classification in 2021, spinal tumors also required genetic studies with some tumors renamed by genetics and methylation profile [3]. The tumor names were changed into spinal ependymoma (SP-EPN) and spinal subependymoma (SP-subEPN) by adding location, and grades should be written in Arabic rather than Roman characters [4]. Here, authors have summarized the updated knowledge of pathological classification of the primary IM-SCTs.
This article describes an updated classification of IM tumors with frequency, age distribution, sex difference, pathological, and genetic hallmarks according to the 5th edition of WHO classification. Authors reviewed primary spinal cord tumors in the archives of SNUH (Table 1). For the past 10 years (2012–2021), and 329 primary IM-SCTs out of 1,765 cases of both EM- and IM-SCTs were reviewed to determine the above mentioned parameters. Intradural EM-SCT, such as schwannoma, meningioma, neurofibroma, malignant peripheral nerve sheath tumor, solitary fibrous tumor, chordoma, metastatic tumor, and vascular malformation, were excluded. Although genetic studies were not conducted in all SNUH cases, most of the genetic abnormalities screened were meaningful in making a diagnosis or predicting patient’s prognosis. Since this article is a case-based review, the authors have added genetic features to the table, but no statistical analysis has been performed. This review of electronic medical records and digital pathology images was approved by the Institutional Review Board of SNUH (IRB No: 2202-097-1301).

Epidemiology of the intramedullary spinal cord tumors of SNUH cases, which are listed by their frequency in the spinal cord
Among primary IM tumors at SNUH, spinal ependymomas (SP-EPN) (n=140, 8.4% of the primary EM- and IM-SCTs and 45.1% of IM-SCT), hemangioblastoma (n=66, 3.7% of primary spinal tumors and 20.1% of IM-SCT), myxopapillary EPN (2% of primary spinal tumors and 11.0% of IM-SCT), and astrocytic tumors (3.1% of primary spinal tumors, 8.8% of IM-SCT), including low-grade (n=8, 14% of spinal astrocytic tumors) and high-grade (n=34, 89.7% of spinal astrocytic tumors), were the most common. The latter high-grade astrocytic tumors included glioblastoma (GBM) IDH-wildtype CNS WHO grade 4 (n=21, 36.8% of spinal astrocytic tumors) and diffuse midline glioma (DMG) H3 K27M-altered (n =13, 22.8% of spinal astrocytic tumors). Spinal subEPN (0.6% of primary spinal tumors, 3.0% of the IM-SCT), ganglioglioma (0.3% of primary spinal tumors, 1.5% of IM-SCT), diffuse leptomeningeal glioneuronal tumors (DLGNT), and atypical teratoid/rhabdoid tumor (AT/RT) (0.1% of primary spinal tumors, 0.3% of IM-SCT, each) were rare (Table 1).
The main classification of EM-SPT remains unchanged in the 2021 new WHO classification, but there are newly introduced rare types of CNS mesenchymal tumors, such as intracranial mesenchymal tumor, FET family gene, composed of fused in sarcoma, the Ewing sarcoma, and the TATA-binding protein-associated factor 2N: cyclic AMP responsive element binding protein 1 (CREB) FET:CREB fusion-positive, capicua transcriptional repressor CIC-rearranged sarcoma, primary intracranial sarcoma, and DICER1-mutant, which can also occur as an EM-SPT [5-8]. In order of increasing frequency, the types of EM-SPT are schwannoma, meningioma, neurofibroma, paraganglioma, chordoma, malignant peripheral nerve sheath tumors, melanocytic tumors, and metastatic tumors [9]. These EM-SPTs are not discussed in this article.
This article summarizes the updated classification of IM-SCTs based on their pathological features and molecular genetic profiles in SNUH cases.
SPINAL EPENDYMOMAS
SP-EPN is the most common glial tumor of the spinal cord and arises from the ependymal cells of the spinal canal [10]. SP-EPNs were 2.6 times more common (n=140, 42.7% of IM-SCT) than astrocytic tumors (n=57, 17.4% of IM-SCT) at SNUH for 10 years from 2012. The incidence of SP-EPNs, myxopapillary EPN, and spinal subEPN comprised 42.7%, 11.0%, and 3.0% of IM-SCT, respectively, at SNUH. The CNS WHO grade 3 SP-EPN was rare, found in 0.5% of primary spinal tumors, 2.4% of IM-SCT, and 5.4% of SP-EPN. The common age for SP-EPN was middle-aged (median, 44 years; range, 2–73 years) with a slight female predominance (male:female ratio 1:1.1) (Table 1). Among the SNUH cases, the most common sites for SP-EPN were at the level of the cervical, lumbar, and thoracic (8:3:1).
SP-EPNs are usually well-circumscribed tumors and have typical perivascular pseudorosettes consisting of a central blood vessel and a surrounding anuclear fibrillary zone (Fig. 1) or true ependymal rosettes with lumina. Tumor cells are monotonous with uniformly round to oval nuclei and salt-and-pepper chromatin. The nucleoli are usually inconspicuous. CNS WHO grade 2 SP-EPNs have a low rate of mitosis rates and a low proliferation index, but necrosis may be present. Rarely, papillary and tanycytic subtypes are observed in the spinal cord. Tanycytic EPNs favor the spinal cord over the intracranium; however, they were not mentioned in the 2021 WHO classification. Tanycytic EPN shows an astrocytoma-like fascicular appearance with indistinct perivascular pseudorosettes, but intratumoral hemorrhage is common (Fig. 2). CNS WHO grade 3 SP-EPNs exhibit high cellularity and brisk mitosis (≥ 20/10 high-power field) with microvascular proliferation, but nuclear pleomorphism is not obvious (Fig. 2). Invasion to the spinal cord parenchyma can occur in CNS WHO grade 3 EPN.
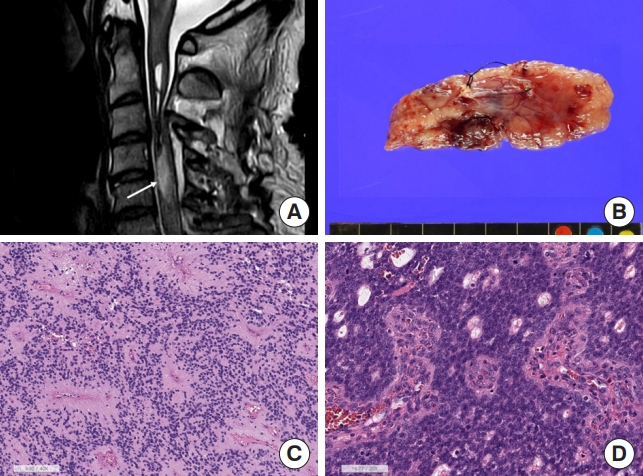
(A) Magnetic resonance imaging shows about 1.7 cm×0.8 cm × 1.9 cm, mildly enhancing intramedullary mass (arrow) in the C4/5 level with syrinx formation from C2 to C5/6 level with fluid-fluid level, suggestive of hemorrhage and cord edema from C1 to C5 level. (B) Grossly this ependymoma is a well-demarcated finely lobulated tumor. (C) Spinal ependymoma central nervous system (CNS) World Health Organization (WHO) grade 2 shows perivascular pseudorosettes with monotonous small round cells and small round nuclei (H&E; scale, 100 μm). (D) Spinal ependymoma CNS WHO grade 3 reveals high cellular hyperchromatic nuclei with perivascular pseudorosettes and microvascular proliferation (H&E; scale, 100 μm).
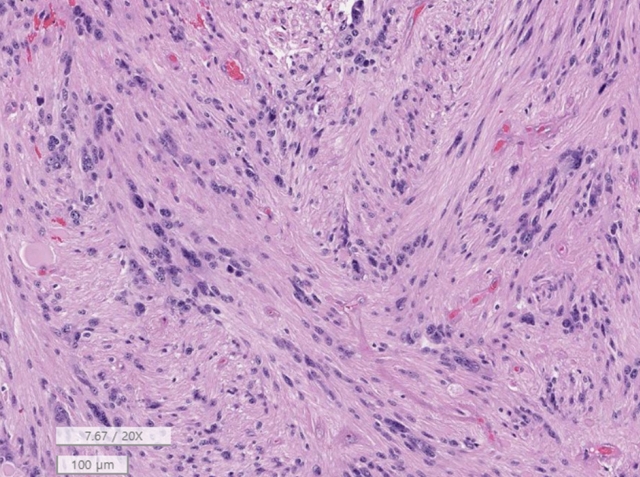
Tanycytic ependymoma shows a fascicular pattern of the tumor with fibrillary cytoplasm of tumor cells and indistinct ependymal rosettes, morphologically indistinct from astrocytic tumors (H&E; scale, 100 μm).
All types of EPNs are positive for glial fibrillary acidic protein (GFAP), epithelial membrane antigen (EMA), S100 protein, and vimentin [11]. GFAPs are usually more accentuated in the perivascular anucleated fibrillary zone but diffuse positivity is not uncommon. EMA positivity is represented by a dot-like or tiny ring-like appearance, which is an ultrastructural intracytoplasmic microrosette with microvilli and cilia. They are generally negative for oligodendrocyte transcription factor 2 (Olig2) and synaptophysin [11], and these markers are helpful for the differential diagnosis of astrocytic or neuronal tumors. Since SP-EPN does not show EZHIP overexpression or H3K27me3 loss in the immunohistochemical study, the presence of these 2 findings should first rule out the drop-down metastasis of posterior fossa group A-EPN. However, ZFTA-RELA fusion-positive primary SP-EPN has been reported [12].
Although SP-EPNs are morphologically similar to supratentorial and posterior fossa EPNs, the molecular genetics and methylation profiles of these SP-EPNs are different from those of intracranial EPNs [13,14]. The most common genetic abnormalities of SP-EPNs are one copy loss of NF2 or NF2 mutations [11,15,16]. CNS WHO grade 3 EPNs commonly have multiple chromosomal copy number aberrations, in addition to one copy loss of NF2 or NF2 mutations. According to Lee et al. [17] the frequency of NF2 mutations in spinal and intracranial EPN was 32.1 and 4.4%, respectively.
MYCN gene amplified SP-EPN (SP-EPN-MYCN) has been recognized as a rare subtype of SP-EPN characterized by multiple tumors and aggressive behavior [18]. This SP-EPN-MYCN has histopathological features of high-grade ependymoma, such as high cellularity, microvascular proliferation, brisk mitosis, tumor necrosis, and high MIB-1 proliferation index. Robust nuclear MYCN expression or in situ hybridization with the MYCN-locus probe may be useful for detecting MYCN amplification, as well as NGS studies (Fig. 1D).
MYXOPAPILLARY EPENDYMOMAS
Myxopapillary EPNs, CNS WHO grade 2, are not uncommon, constituting approximately 11% of IM-SCTs in SNUH. These tumors commonly occur at the distal thoracic to lumbar region (T12 to L2, 3), including the sacrum and filum terminale, but rarely at the upper thoracic or cervical levels of the spinal cord [19]. The median age of SNUH patients with myxopapillary EPNs was 40 years (range, 15–80 years). Grossly, these tumors are well-encapsulated and often dumbbell-shaped solid masses composed of hyalinized blood vessels and a myxoid or mucinous intercellular matrix. The tumor cells show a monotonous polygonal appearance but sometimes long bipolar fibrillary cytoplasmic processes. The nuclei are usually round to oval and bland-looking, but sometimes enlarged nuclei is present due to degenerating atypia (Fig. 3). These tumors have a favorable prognosis with 10-year overall survival rates > 90% [20]. Myxopapillary EPNs rarely metastasize to extraneural sites [21].
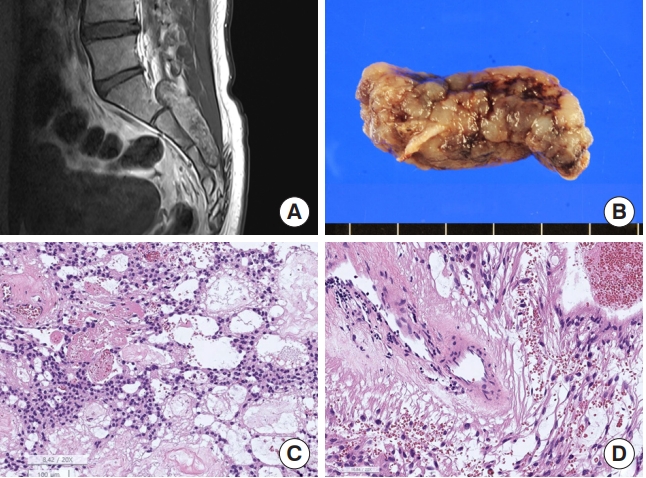
Myxopapillary ependymoma. (A) Magnetic resonance imaging. A 6.1 cm × 1.8 cm × 7.8 cm, T2 heterogeneous hyperintense and hypointense extradural (epidural) soft tissue mass in the S1–3 sacral central canal. There are multifocal pressure bone erosion and resultant central canal widening. (B) The tumor shows a well-demarcated and multilobulated appearance. (C) The tumor shows sheets of polygonal cells with a lake of myxoid and mucinous material and hemorrhage and fibrinous material (H&E; scale, 100 μm). (D) The vascular wall is hyalinized and tumor cells have long fibrillary cytoplasmic processes (H&E; scale, 50 μm).
SUBEPENDYMOMA
SubEPNs are rare primary benign IM-SCTs classified as CNS WHO grade 1 tumors. In SNUH, they account for 0.6% of SPT and 3.0% of IM-SCT and usually occurred between 20 and 60 years (median, 38 years; range, 21–57 years). They commonly occur at cervical and thoracic levels. SP-subEPNs are well-circumscribed or well-encapsulated tumors that show typical microscopic features, including alternative cellular and acellular areas with microrosette-like multiple cell aggregates (Fig. 4). Metastases are extremely rare, and neither necrosis nor spinal cord invasion are observed. Degenerative nuclear atypia is rarely found, but is not a high-grade feature. Occasionally, in otherwise typical cases, fibrillary astroglial or gemistocytic cells may appear (Fig. 4). Immunohistochemical findings are similar to those of EPN; thus, GFAP is diffusely positive and might exhibit focal dot-like positivity for EMA, but negative for synaptophysin and Olig2. Although little is known about the genetic alterations in subEPN, mutations in the TRS1 gene have been found in familial subEPNs [22]. BRAF and H3F3A mutations are absent and H3K27me3 is retained in the tumor cells. Spinal subEPNs are also morphologically identical to intracranial subEPNs; however, the methylation profile of spinal subEPN is different from that of intracranial and posterior fossa subEPN. Even after partial resection, the prognosis is excellent, and recurrence after surgery is rare [23].
Subependymoma. (A) Magnetic resonance imaging. Diffuse intramedullary T2 hyperintensity lesion with swelling of the spinal cord, C6–T5 level, and focal enhancement. (B) Resected tumor shows well-demarcated lobulated yellow tancolored tumor. (C) Histopathologically alternative cellular and acellular areas with nuclear aggregates (H&E; scale, 50 μm). (D) Some ependymomas show gemistocytes-like plump eosinophilic cytoplasm (H&E; scale, 50 μm).
DIFFUSE ASTROCYTIC TUMORS, INCLUDING ADULT-TYPE DIFFUSE GLIOMAS AND PEDIATRIC-TYPE DIFFUSE GLIOMAS
Astrocytomas constituted 17.4% of IM-SCT, including diffuse astrocytoma (n=29, including 8 low- and 21 high-grade astrocytomas), DMG H3K27-altered (n=13), and pilocytic astrocytoma (n=15). Pilocytic astrocytoma, CNS WHO grade 1 was found in 4.6% of IM-SCT, diffuse low-grade gliomas (2.4% of IM-SCT), and diffuse high-grade glioma (DHGG, 10.4% of IM-SCT). In SNUH cases, GBM IDH-wildtype (6.4% of IM-SCT) and DMG H3 K27M-altered (4.0% of IM-SCT) were the most common malignant astrocytic tumors (DHGG).
According to Hamilton et al. [24] only 13% of spinal gliomas, including pediatric gliomas, was malignant. As the histopathology of spinal astrocytic tumors is similar to that of intracranial astrocytic tumors (Fig. 5), low grade gliomas (LGGs) have scant mitoses, no microvascular proliferation, no necrosis and low Ki-67 labeling index (Fig. 5B). GBM usually had pleomorphic nuclei of tumor cells, microvascular proliferation, and/or necrosis (Fig. 5A, C).

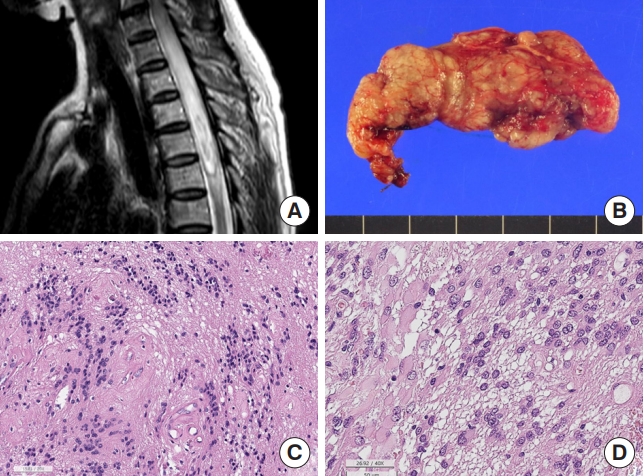
Spinal astrocytoma. (A) Magnetic resonance imaging. Intramedullary glioblastoma shows T2 heterogeneous mass (arrow) at T7–12 levels with multifocal enhancement and is associated with syrinx from C4 to T7 level. (B) The low-grade astrocytic tumor shows sheets of astrocytic cells with eosinophilic cytoplasm (H&E; scale, 50 μm). (C) The highly cellular tumor has multiple foci of necrosis (asterisk) with pseudopalisading nuclei (H&E; scale, 100 μm). (D) Diffuse midline glioma shows sheets of hyperchromatic elongated cells. Left: the tumor cells of DMG, H3K27-altered are arranged in sheet with elongated nuclei, looked like astrocytic tumor (H&E of DMG H3K27-altered; scale, 50 μm); right: the tumor cell nuclei of DMG are positive for K27M (K27M immunohistochemistry; scale, 50 μm).
The genetics of spinal LGG is not well known because they are rare, but BRAF gene alterations have been reported [25]. Most spinal GBMs were IDH-wildtype de novo tumors with EGFR amplification and/or PTEN or CDKN2A homozygous deletion, and TERT promoter and/or TP53 mutations have been found in the SNUH series, which is similar to the cases of Nagaishi et al. [26].
Although several cases of spinal IDH-mutant astrocytomas and spinal oligodendrogliomas have been reported, they are extremely rare [27,28]. Hemispheric gliomas, such as diffuse hemispheric glioma, H3 G34-mutant, and infant-type hemispheric glioma, are also extremely rare.
DMG, H3 K27-altered in the spinal cord, is a relatively recently recognized aggressive glioma classified as CNS WHO grade 4 [29]. Since the median age of DMG H3 K27-altered was 32.5 years old (range, 19–75 years old) in SNUH cases, spinal DMG can occur at any age and is more common in adults. These tumors carry somatic mutations of H3F3A or HIST1H3B/C [29]. Morphologically, DMG can have various grades of astrocytic tumors (Fig. 5D); therefore, it can appear as low-grade astrocytoma or typical GBM, or have a primitive neuroectodermal tumor-like appearance. The tumor cells were positive for GFAP, and the tumor cell nuclei were positive for the H3K27M-mutant specific antibody, K27M (Fig. 5D). However, spinal cord DMG H3 K27-altered has a slightly better prognosis than spinal GBM, CNS WHO grade 4 [30]. TP53, ATRX, and ACVR1 mutations commonly accompany these tumors [31]. However, EGFR-mutant DMG, known as bithalamic glioma, and EZHIP-overexpressing spinal DMG have never been reported in the spinal cord.
CIRCUMSCRIBED ASTROCYTIC GLIOMAS
Pilocytic astrocytoma is a relatively well-circumscribed and indolent CNS WHO grade 1 astrocytoma. Although pilocytic astrocytoma commonly occurs in the posterior fossa and optic pathway in children and adolescents, spinal pilocytic astrocytoma accounted for 4.3% of all IM-spinal tumors at SNUH (Table 1). The age of onset of spinal pilocytic astrocytomas was slightly higher (median, 37 years; range, 1–65 years) than that of supratentorial tumors.
The histopathology of spinal pilocytic astrocytoma is similar to that of intracranial pilocytic astrocytoma, showing low cellularity and bland-looking elongated nuclei with bipolar cytoplasmic processes. Vascular hyalinization and rosenthal fibers are common (Fig. 6). Occasionally, degenerative nuclear atypia is observed.
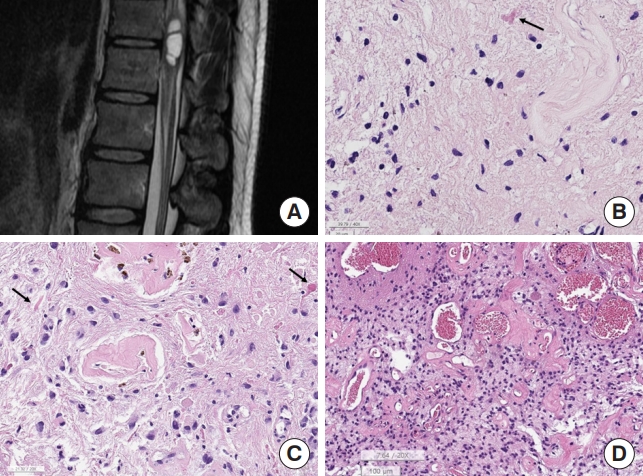
Spinal pilocytic astrocytoma, 3 cases with various histology. (A) Magnetic resonance imaging. About bout 3.2-cmsized dorsally exophytic intramedullary tumor at the C1–2 level and shows T2 high signal intensity. (B) The tumor shows low cellularity, a hyalinized blood vessel, and a rosenthal fiber (arrow) (H&E; scale, 20 μm). (C) Another pilocytic astrocytoma shows nuclear pleomorphism which is degenerated nuclear atypia with hyalinized blood vessels and rosenthal fibers (arrows) (H&E; scale, 50 μm). (D) The other pilocytic astrocytoma shows moderately cellularity with ectatic and congested blood vessels and hyalinized vascular wall (H&E; scale, 100 μm).
Ninety percent of posterior fossa-pilocytic astrocytomas and 60% of optic pathway-pilocytic astrocytomas are known to have KIAA1549:BRAF fusion, and the remaining cases have mitogen-activated protein kinase (MAPK) pathway gene alterations, including BRAF V600E, NF1, PTPN11, and FGFR1 mutations [32]. Although 40% of spinal pilocytic astrocytomas have KIAA1549: BRAF fusion, BRAF V600E mutation has been found in 4% of spinal pilocytic astrocytomas. Furthermore, homozygous deletion of CDKN2A is slightly more common in spinal cord and brainstem pilocytic astrocytoma than in cerebellar ones (21.1% vs. 33.3%) [33]. FGFR1:TACC1 fusion has been reported in pilocytic astrocytoma occurring in the brainstem and near full-length of the cervical spinal cord of a 22-year-old female [34]. One of our spinal pilocytic astrocytomas in a 65-year-old male had an FGFR1: TACC1 fusion.
Other circumscribed astrocytic gliomas, such as high-grade astrocytoma with piloid features, pleomorphic xanthoastrocytoma, and MN1-altered astroblastoma, rarely occur in the IM-spinal cord; however, subependymal giant cell astrocytoma and chordoid glioma have never been reported in the spinal cord [35-38].
GLIONEURONAL AND NEURONAL TUMORS
Ganglioglioma, CNS WHO grade 1 was the most common glioma, accounting for 1.5% of primary IM-SCT in SNUH. DLGNT rarely occurred in the spinal cord (0.3% of IM-SCT).
Ganglioglioma is a relatively well-demarcated, slow-growing neoplasm of childhood. The median age of patients with ganglionma was 5 years old (range, 2–10 years) in SNUH. Sometimes, they involve the long segments of the spinal cord [39]. Gangliogliomas are composed of 2 cell components, neoplastic ganglion cells and glial cells (Fig. 7). These tumors are usually caused by alterations in the MAPK signaling pathway, usually with a KIAA1549:BRAF fusion; however, BRAF V600E and NF1 (sometimes biallelic) mutations or deletions have also been observed. Very rarely, spinal gangliogliomas contain only H3K27M mutations.

Spinal ganglioglioma. (A) Magnetic resonance imaging reveals intramedullary, bulging mass (arrows) involving C6–T5 spinal cord - eccentric location and T2 high SI with partial enhancement. (B) The tumor is composed of mature ganglion cells and glial cells (H&E; scale, 100 μm). (C) The glial cells are positive for CD34 but ganglion cells are negative for CD34 (scale, 50 μm). (D) The glial cells are positive for glial fibrillary acidic protein (GFAP), but ganglion cells are negative for GFAP (50 μm).
DLGNT is a low-grade glioneuronal neoplasm characterized by widespread diffuse involvement of the leptomeninges and superficial brain parenchyma by monotonous oligodendroglioma-like cells with bidirectional differentiation [40]. These tumors are commonly found in the subpial region of the basal surface of the brain, brainstem, and spinal cord. Genetically, KIAA1549: BRAF fusion was found in 72% of the studied cases while 1p and/or 19q deletion was found in other cases [41]. 1p/19q codeletion has been identified in 18%–33% [41,42]. Based on the methylation profiles, these tumors are classified into methylation classes 1 and 2 (MC1 and MC2). Although MC1 is roughly similar to CNS WHO grade 2 gliomas in clinical course, MC2 has anaplastic features, 1q gain, and/or a worse prognosis than MC1 [43].
CNS EMBRYONAL TUMORS
Among embryonal tumors, AT/RT, CNS WHO grade 4, rarely occur in the IM-spinal cord of infants (0.6% of IM-SCT), while other embryonal tumors are extremely rare. Approximately 7.6% of AT/RT occurs in the spinal cord [44]. If this tumor develops in the EM-spinal cord, i.e., extradural or paravertebral, or in patients aged 3 years or older, metastasis of extracranial malignant rhabdoid tumor should be ruled out first. The pathological and genetic features were identical to those of intracranial AT/RT in SNUH. The tumor presents as a monotonous small round cell tumor with an eccentric nucleus and a prominent eosinophilic rhabdoid appearance, but primitive small round cells with scanty cytoplasm can be seen (Fig. 8). Tumor cell nuclei are usually negative for INI-1 and the Ki-67 labeling index is usually very high. SMARCB1 mutations or homozygous deletions are the genetic hallmarks of this tumor [45]. Since poorly differentiated chordomas also have SMARCB1/INI-1 loss, pathologists should keep it in mind when making a differential diagnosis [46]. Biological behavior is as aggressive as intracranial AT/RT [45].
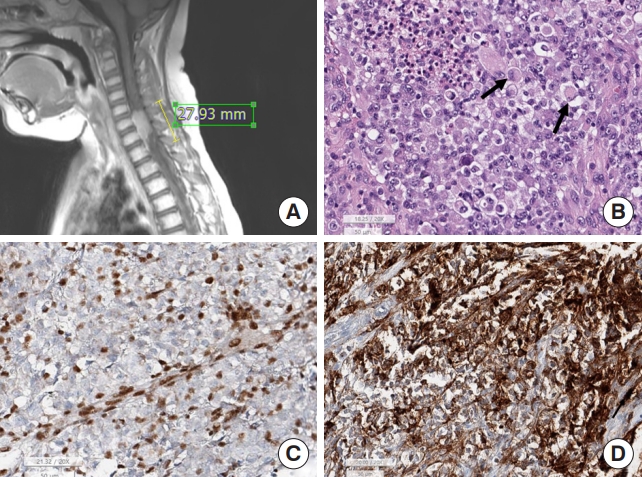
Atypical teratoid rhabdoid tumor. (A) Magnetic resonance imaging shows a 2.8-cm elongated epidural enhancing mass between C5 and T1. (B) Light microscopically the tumor shows small round cells with eccentrically located nuclei and eosinophilic cytoplasm (arrows), which is a rhabdoid feature. (C) The tumor cell nuclei are negative for INI-1, but include internal control, such as endothelial cells and some inflammatory cells are positive for INI-1 (INI-1 immunohistochemistry; scale, 50 μm). (D) The tumor cells are at least focal positive for epithelial membrane antigen (EMA; scale, 50 μm).
GERM CELL TUMORS
Mature cystic teratomas, immature teratomas, and pure germinomas can occur in the spinal cord. In the SNUH series, these germ cell tumors are extremely rare, with each tumor subtype occurring in 0.1% of SPT and 0.6% of IM-SCT. Germinoma occurred in T12–L1 and young adults, and interestingly, 2 cases of SNUH germinoma occurred in 22-year-old females. Teratomas also commonly arise in the lower spinal level from L2 to the coccyx at any age. The 2 SNUH teratomas occurred in an infant and a 56-year-old woman. The histopathology of spinal teratomas and germinomas is identical to that of extraspinal tumors. Pure germinomas consist of malignant germ cells and lymphoplasma cells. Malignant germ cells are arranged in sheets of polygonal-shaped malignant germ cells, which have centrally located rounded nuclei and prominent nucleoli (Fig. 9). The cytoplasm is moderate to plump and shows a pink to clear appearance because of the large amount of glycogen and lipid vacuoles. High rates of mitosis and necrosis are also common. Characteristically, germinoma cells have a positive membranous expression of c-kit and are commonly positive for placental alkaline phosphatase. Mature teratomas have mature 3-germ-layer tissues. Immature teratomas contain primitive neuroepithelial tubules. Genetically, one germinoma of SNUH cases had an ALK gene mutation (p.Gly926fs, c.2775delAinsGG); in addition, KIT mutation and chromosome 12p gain or amplification have been reported in intracranial germ cell tumors [47].
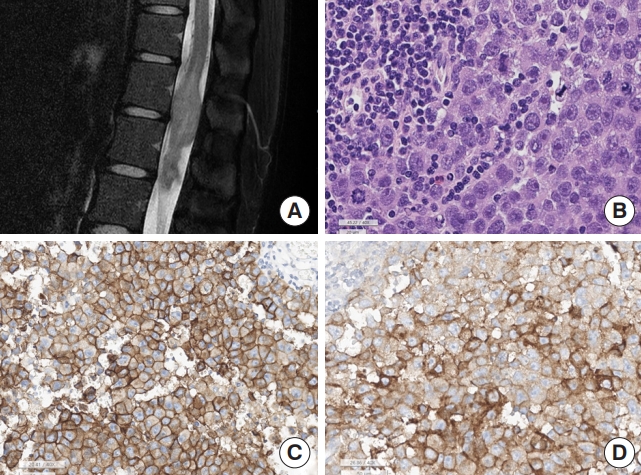
Germinoma. (A) Magnetic resonance imaging shows T2 heterogeneous intensity mass at T12–L1 spinal cord, involving conus medullaris. (B) The tumor is composed of biphasic, malignant germ cells and lymphoplasma cells (H&E; scale, 20 μm). The malignant germ cells show large round nuclei and prominent nucleoli. There are frequent mitoses. (C) The tumor cell membrane is positive for c-kit (scale, 50 μm), and (D) placental alkaline phosphatase (PLAP; scale, 50 μm).
HEMANGIOBLASTOMAS
Hemangioblastoma, CNS WHO grade 1, is the second most common IM-SCT after ependymomas, accounting for 3.7% of all primary SPT and approximately 20.0% of IM-SCT in SNUH. Hemangioblastomas do not undergo malignant transformations. However, both IM and EMhemangioblastomas have been reported [48,49]. In SNUH cases, hemangioblastoma was 1.5 times more common than astrocytic tumors and 1/3 less common than SP-EPN. Hemangioblastomas are adult tumors that occur in a wide range of patients (mean age, 43 years; range, 16–81 years). The male to female ratio was 2:1. Hemangioblastomas occur at any level from the cervical spine to the lumbar spinal cord. The tumors are well-circumscribed, pseudoencapsulated, and capillary-rich solid tumors. The tumor cells are stromal cells with foamy cytoplasm, and the capillaries are nonneoplastic components (Fig. 10). Ultrastructurally, tumor cells have many cytoplasmic fat vacuoles and intermediate filaments, which produce foamy cytoplasm on H&E staining. Tumor cells express NSE, S100, alpha-inhibin, D2-40, and brachyury (cytoplasmic expression), which can help in the differential diagnosis [50]. The pathogenesis and cells of origin remain unknown. In rare cases, hyaline globules are present. This tumor may be sporadic or be associated with von Hippel-Lindau disease. However, the tumor usually has various kinds of VHL gene mutations, such as missense, slicing, insertion, or deletion mutations [51,52]. When the tumor is completely removed, the prognosis is good [53].
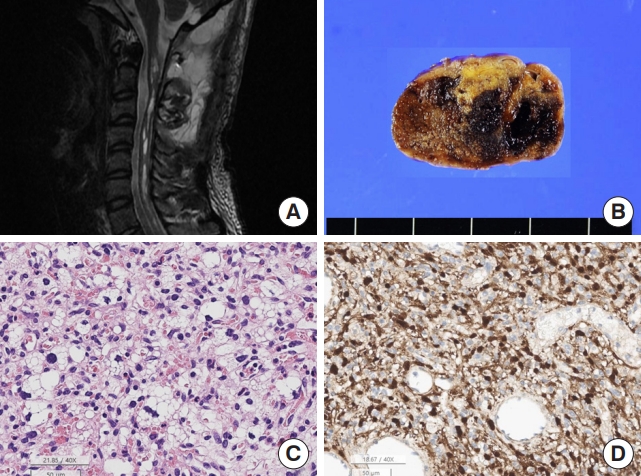
Hemangioblastoma. (A) Magnetic resonance imaging shows a 5.1-cm solid and cystic tumor with multifocal enhancement in at C2–4 Spinal cord with a tiny intramedullary enhancing nodule at C2–T3 levels with spinal cord edema. (B) The tumor shows a well-demarcated hemorrhage mass. (C) The tumor is highly vascular with rich capillaries. The tumor cells have round to mildly pleomorphic nuclei and foamy cytoplasm (H&E; scale, 50 μm). (D) The nuclei and cytoplasm of the tumor cells are positive for S-100 protein, but the capillary endothelial cells are negative for S-100 protein (S-100 immunohistochemistry; scale, 50 μm).
CONCLUSION
Theoretically, any type of primary CNS tumor can occur in the intramedullary spinal cord; however, the most common IM-SCT is EPN, followed by hemangioblastoma and astrocytoma. SP-EPN and SP-subEPN are morphologically identical to those of the supratentorial or posterior fossa, but their molecular genetic and methylation profiles differ from those of intracranial EPN. Therefore, these tumors should have ‘spinal’ in the tumor name. DLGNT can arise in the spinal cord and are characterized by a KIAA1549:BRAF fusion, 1p and/or 19q deletion, or 19q gain. In addition, some spinal pilocytic astrocytomas are characterized by FGFR1:TACC1 fusion, in addition to alterations in the MAPK pathway. Myxopapillary ependymomas characteristically occur in the lumbosacral area and are regarded as CNS WHO grade 2 [4]. Although spinal DMG H3 K27M-altered is a high-grade glioma, the biological behavior of spinal DMG is better than that of spinal GBM IDH-wildtype, CNS WHO grade 4. Rarely, pilocytic astrocytoma, ganglioglioma, diffuse leptomeningeal glioma, and atypical teratoid rhabdoid tumors occur in IM-SCT; they may share a KIAA1549:BRAF fusion. However, although several cases of spinal IDH-mutant astrocytomas have been reported, hemispheric gliomas (such as diffuse hemispheric glioma, H3 G34-mutant, and infant-type hemispheric glioma) or IDH-mutant gliomas (including IDH-mutant astrocytoma and oligodendroglioma) are extremely rare.
Notes
Ethics Statement
The institutional review board of our hospital approved this study (IRB No: 2202-097-1301) and has therefore been performed under the ethical standards set out in the 1964 Declaration of Helsinki and its subsequent amendments. As this study is a retrospective review of anonymized electronic medical records, pathology, and results of NGS data utilizing a brain tumor-specific somatic gene panel, informed consent was waived from our IRB under the Korean Bioethics and Safety Act. All materials had been obtained for the electronic medical record of the patients, which were anonymized and retrospectively reviewed. No extra-human materials were obtained from the patients for this study. Under the Korean Bioethics and Safety Act, additional consent to publish was waivered.
Conflict of Interest
The authors have nothing to disclose.
Funding/Support
This study was supported by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI14C1277).
Author Contribution
Conceptualization: SP; Data curation: JKW, CHK, JHP, SK, SC, CKC; Writing - original draft: SP; Writing - review & editing: SP