INTRODUCTION
Developmental bony craniovertebral junction (CVJ) anomalies frequently occur with neural abnormalities, indicating an embryologic relationship. They appear to be the result of maldevelopment of the cartilaginous neural cranium and adjacent vertebral skeleton during the early embryonic weeks [1,2]. The spectrum of congenital/developmental anomalies consists of atlantoaxial dislocation (AAD), and basilar invagination (BI), with occipitalization of C1 arch. Besides, there may be the presence of Os odontoideum, Chiari malformation (with or without syringomyelia), Klippel Feil anomaly, Clival segmentation anomaly. Other uncommon anomalies include proatlas segmentation failure, condylar hypoplasia, bifid C1 arch, hemivertebrae. Besides, most of the patients we see have phenotypic markers (e.g., low set ears, hyper-extendibility, etc., see below). We frequently see developmental CVJ cases in the Indian subcontinent. We have used the term ‘developmental’ to indicate that even though the underlying pathology is congenital, the symptoms develop much later, usually the first or second decade of life.
The anomalies of the CVJ are associated with several connective tissue disorders [3-7], and familial basis is also well documented [8-13] in the literature. As in most autosomal dominant disorders, we find a varied expression of clinical and morphological anomalies in congenital CVJ anomalies [14]. Earlier studies demonstrated an autosomal dominant trait for familial CVJ [15,16] (especially basilar impression) [15,17]. Overall, many authors have viewed CVJ as a single autosomal dominant disorder expressed in a variable sequence as osseous malformations and Chiari malformation [14].
More recently, we have demonstrated that deformed C1–2 joints may produce a mechanical disadvantage that itself may lead to a progressive deformity [18-26]. Probably, weight-bearing over a time coupled with age-related degenerative changes may precipitate BI and subsequently cause clinical features. Thus, many patients with congenital CVJ present with clinical symptoms present between first to the second decade of life and not during childhood.
Despite this evidence, there are very few studies till date, which have attempted to study developmental CVJ anomalies' genetic basis [25,27]. We found that most studies were based on the Caucasian population on genome-wide association (GWAS) [28] and tagged SNP’s [29]. Boyles et al. [28] in a GWAS, screened genome-wide linkages with 71 affected individuals and analyzed over 10,000 single-nucleotide polymorphisms (SNP) across the genome and found linkage to regions on chromosome 9 and chromosome 15, at loci 15q21.1-q22.3 and 9q22.31. They found the fibrillin1 gene (FBN1) on chromosome 15, as the most biologically plausible gene for Chiari malformation I (CMI). FBN1 is known to encode for the gene for fibrillin. Three different genes FBN1, FBN2, and FBN3 encodes fibrillins in humans. Fibrillins are large (~350,000 MW) structural macromolecules that not only contribute to the integrity and function of all connective tissues but also target and sequester members of the transforming growth factor-β superfamily of growth factors and contribute to organ formation and repair [30]. Thus mutations in fibrillins can contribute effectively to tissue growth and homeostasis. Each fibrillin molecule contains 47 epidermal growth factor (EGF)-like domains, 43 are predicted to bind to calcium (cbEGF), 7 8-cysteine containing domains (8-cys), 2 “hybrid” domains that share features of both the 8-cysteine domain and the EGF-like domain, a proline-rich domain, and amino- and carboxyl- terminal domains [30]. Genetic studies revealed mutations in the fibrillin molecules as an essential determinant in clinical phenotypes of Marfan syndrome (MFS). Till now, 1,847 different mutations in 3,044 DNA samples have been reported in the FBN1 mutation database (http://www.umd.be/FBN1/- last updated 8/28/2014). Mutations related to atypically severe phenotypes were found to be clustered in exons 24–32 in MFS [31]. This region is composed of a central longest stretch of 12 cbEGF repeats, which is believed to form a rigid rod-like structure with a crucial role in microfibril assembly. Also, genotype-phenotype correlations of mutations in FBN1 are associated with specific clinical features of MFS. The most consistent correlation was found in the middle region of the gene (exons 24–32) with all cases of “neonatal” MFS and its other severe forms [32-34]. Despite these studies, the exact phenotype for any given FBN1 mutation is not predicted so far.
Considering that fibrillin is an important component of microfibrils, we hypothesized that mutations in FBN1 might precipitate BI and AAD by causing loss of structural integrity of ligaments. Since the FBN1 gene is quite large, we planned first to study exons 24–28, which are also hotspots for mutations in MFS. Then we screened the rest of the 60 exons in 20 patients. To best of our knowledge, this is the first of its kind of study involving this category of patients. We also correlated the study with joint morphology study in 100 patients with developmental AAD and BI.
MATERIAL AND METHODS
1. Ethics Statement
The Institute Ethics Committee approved the study. A detailed written consent was taken from both the patients and control samples explaining to them the study’s details and possible outcome.
The main inclusion criteria for the study was the presence of a nonsyndromic structural CVJ anomaly. All patients included in the study had BI with AAD. Besides, the various other radiological features included one or more of these features: occipitalization of C1 arch, clival segmentation anomalies, Chiari malformation, hydrocephalus, and/or os odontoideum. We have not included patients with specific known genetic syndromes in this study e.g., Down syndrome. Similarly, we excluded all patients with traumatic or inflammatory etiologies.
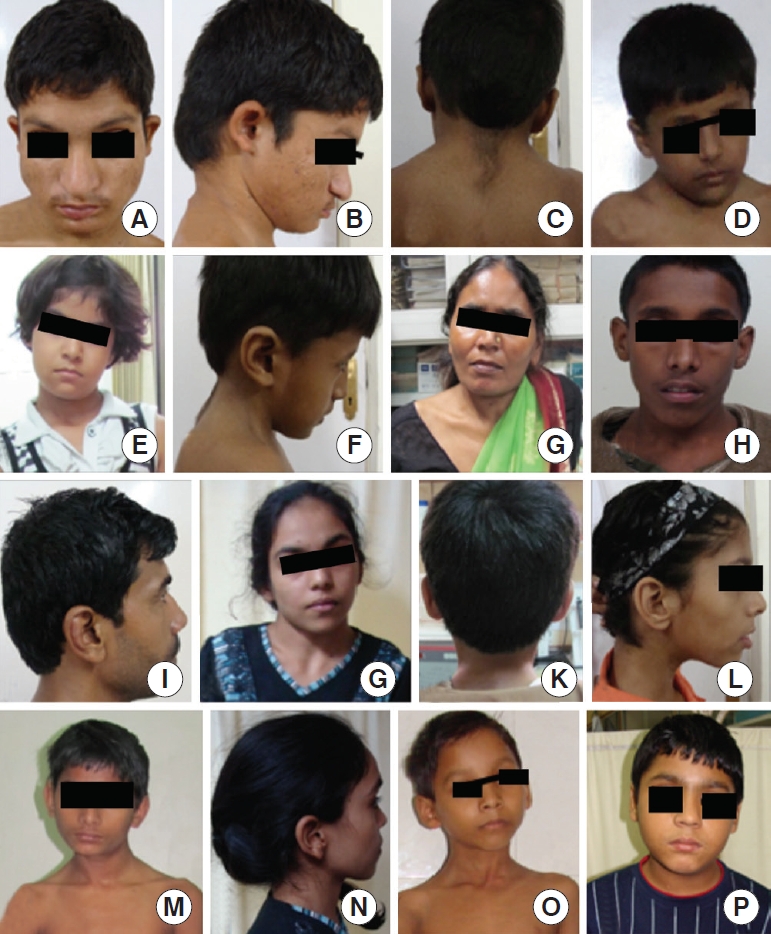
All patients underwent a complete clinical work up, and the phenotypic features carefully noted (Figs. 1, 2). Imaging included magnetic resonance imaging (MRI) (with contrast) of the CVJ with whole spine screening and a computed tomography (CT) scan. The CT scan consisted of thin-slice bone windows with sagittal and coronal reconstruction.
The control included healthy subjects, who had attended the outpatient department with trivial noncongenital problems (e.g., degenerative neck or back problem) and consented to give their blood samples. All the subjects included in the study were from the same ethnic Indian population.
2. Collection of Samples for Genetic Studies
Venous blood samples measuring 5 mL were obtained in ethylenediaminetetraacetic acid (EDTA) tubes. Control samples (50 healthy subjects, 100 reference alleles) were taken from the general healthy population presenting to the outpatient department either with trivial noncongenital problems (e.g., degenerative neck or back pain) or were healthy attendants of the patients who consented to provide their blood.
3. DNA Isolation and Quantification
Genomic DNA was extracted using conventional phenolchloroform method [35]. Amplification [36] of 65 exons of FBN1 gene was carried out in a 25-µL reaction volume using 50 ng of genomic DNA as a template and 10 pmol of each primer (Table 1) [37], buffer with a final Mg2+ concentration of 3.7mM, 0.75mM of each dNTP and 0.5U Taq polymerase. The amplifications were carried out using the following cyclic conditions: initial denaturation at 95°C for 10 minutes, followed by 30 cycles at 95°C for 25 seconds, for annealing a range of temperature from 55°C–60°C for 25 seconds were used, followed by 72°C for 25 seconds, and a final extension at 72°C was given for 10 minutes.
1) Mutation screening
The amplified products were screened using single-stranded conformation polymorphism (SSCP)–heteroduplex [37] analysis. Polymerase chain reaction products were denatured in formamide dye (0.01M EDTA, 98% formamide, trace xylene cyanol, and bromophenol blue) and run on 8% polyacrylamide gels (acrylamide 49:1) for 16 hours at 250 V and silver stained. The samples which migrate differently in SSCP–heteroduplex analysis were sequenced using a fluorescent cycle-sequencing kit. Sequencing conditions consisted of initial denaturation for 1 minute at 96°C; 30 cycles of 96°C for 30 seconds, 15 seconds at 50°C, and 60°C for 4 minutes. Bidirectional sequencing was performed for all the samples exhibiting shifts using ABI 3130 genetic analyzer (Applied Biosystems, Alameda, CA, USA). The sequencing results were compared with the original data using NCBI BLAST (Gen Bank Accession No. AC022467 and AC 084757) and typical FBN1 reference sequence (ENST00000316623). All sequence variations were analyzed in both control, and patient samples and the presence of the mutations were reported in terms of percentage.
2) Radiological measurement of joint morphology
The study population comprised of 100 patients with ‘nonsyndromic, developmental bony CVJ anomalies with BI and AAD’ (see below for definition) with an occipitalized C1 arch in the age range of 15 to 45 years old. Patients of < 15 years of age were excluded due to incomplete physiological bony fusion, and patients of > 45 years of age were excluded because of expected degenerative changes [38]. Besides, the following patients were also excluded: (1) traumatic AADs, (2) polytrauma involving other areas of the cervical spine, (3) rheumatoid arthritis, (4) inflammatory pathologies, such as tuberculosis, (5) patients where imaging did not show the clivus or hard palate, and (6) known genetic syndromes, such as Down or Marfan.
(1) Nonsyndromic, developmental CVJ anomalies
These anomalies included various bony irregularities that suggest a congenital origin, including one or more of the following: occipitalization of the C1 arch with or without fusion at other levels, os odontoideum, clival segmentation anomalies, and Chiari malformation. The features of AAD and BI were observed in all patients and formed the primary inclusion criteria for patients. The patients had one or more of the following phenotypic features: low hairline, short neck, low set ears, clinodactyly, increased first interdigital space of the foot, hyperelastic joints, and arachnodactyly. None of the patients had any known genetic syndrome anomalies.
(2) Controls
The control group included an equal number of age and sexmatched subjects who had undergone a CT scan of the cervical spine and CVJ as a part of the screening procedure following minor injuries. We included only those patients without any evidence of trauma to the cervical spine and CVJ.
(3) Radiological studies
All craniometric measurements were performed using CT rather than x-rays and MRI’s because of the better identification of the bony landmarks on the CT scan.
Thin-slice CT scans (0.63 or 0.7 mm) with reconstructed views were used. All scans were obtained on a 64-MDCT scanner (Aquilion 64, Toshiba Medical Systems, Otawara, Japan) using a rotation time of 500 msec, a tube voltage of 120 kV, and a tube current 40–369 mA. The acquired images were reconstructed into 0.63-mm-thick CT images in 3 orthogonal planes on a dedicated workstation using 3-dimentional software (Vitrea 2, Vital Images, Minnetonka, MN, USA). The midsagittal plane was reconstructed by realigning the positioning crosshairs on the axial images. Images were analyzed on a preset bone window setting of a 2700 HU width and a length of 350 HU.
(4) Measurements
The radiological parameters included atlantodental interval (ADI) [39], and Wackenheim clival canal line, Chamberlain line, McRae line, and modified Ranawat’s line were used to measure BI [25,40-44]. All these techniques have been previously described [25].
The following joint dimensions were measured bilaterally. These have been described earlier [20,22-24,26]. These include sagittal and coronal joint inclinations, and craniocervical tilt [20,22-24,26]. The details of these measurements have been shown in Fig. 3.
We performed the data analysis using IBM SPSS Statistics ver. 20.0 (IBM Co., Armonk, NY, USA). We used the independent t-test for assessing the continuous numerical parameters comparing patients and controls. For the correlation between 2 continuous numerical values, we used the Pearson correlation test.
(5) Other investigations
All patients underwent a complete systemic examination. x-ray chest, electrocardiogram, and a cardiological examination was performed to look for any cardiac anomalies. Dual-energy x-ray absorptiometry scans and osteoporosis work was performed when required. Vitamin D3 was performed in all patients. In patients with low Vitamin D3, supplementation was provided before admission. Ultrasound abdomen was performed to look for any anomalies in abdomen and bladder.
RESULTS
1. Clinical Profile
We included 50 cases and compared them with equal number of age and sex-matched controls. The age of the patients ranged from 8–61 years (mean age, 40.6±12.4 years). The clinical features included neck pain (48 cases, 96%), numbness of the limbs (44 cases, 88%), and unsteady gait (43 cases, 86%) (Table 2). The phenotypic features included low set ears (41 cases, 82%), followed by clinodactyly (30 cases, 60%) and increased the first interdigital space of the foot (26 cases, 52%) (Figs. 1, 2). The commonest radiological finding was BI (34 cases, 68%), followed by AAD (31 cases, 62%) and clival segmentation (29 cases, 58%). Statistical comparison of genetic differences with the control group could not be done as all the mutations were observed in 78% of the disease cohort and 0% in the control group. None of the patients in our cohort had any systemic cardiac and skeletal anomalies.
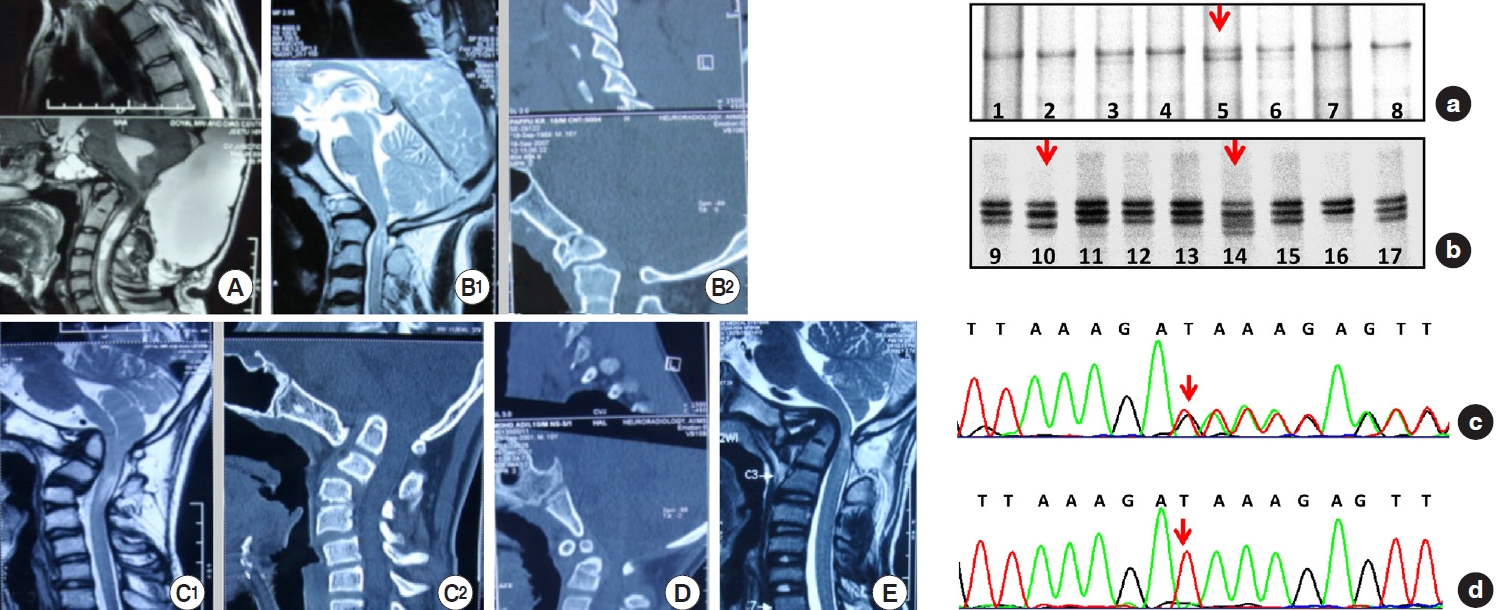
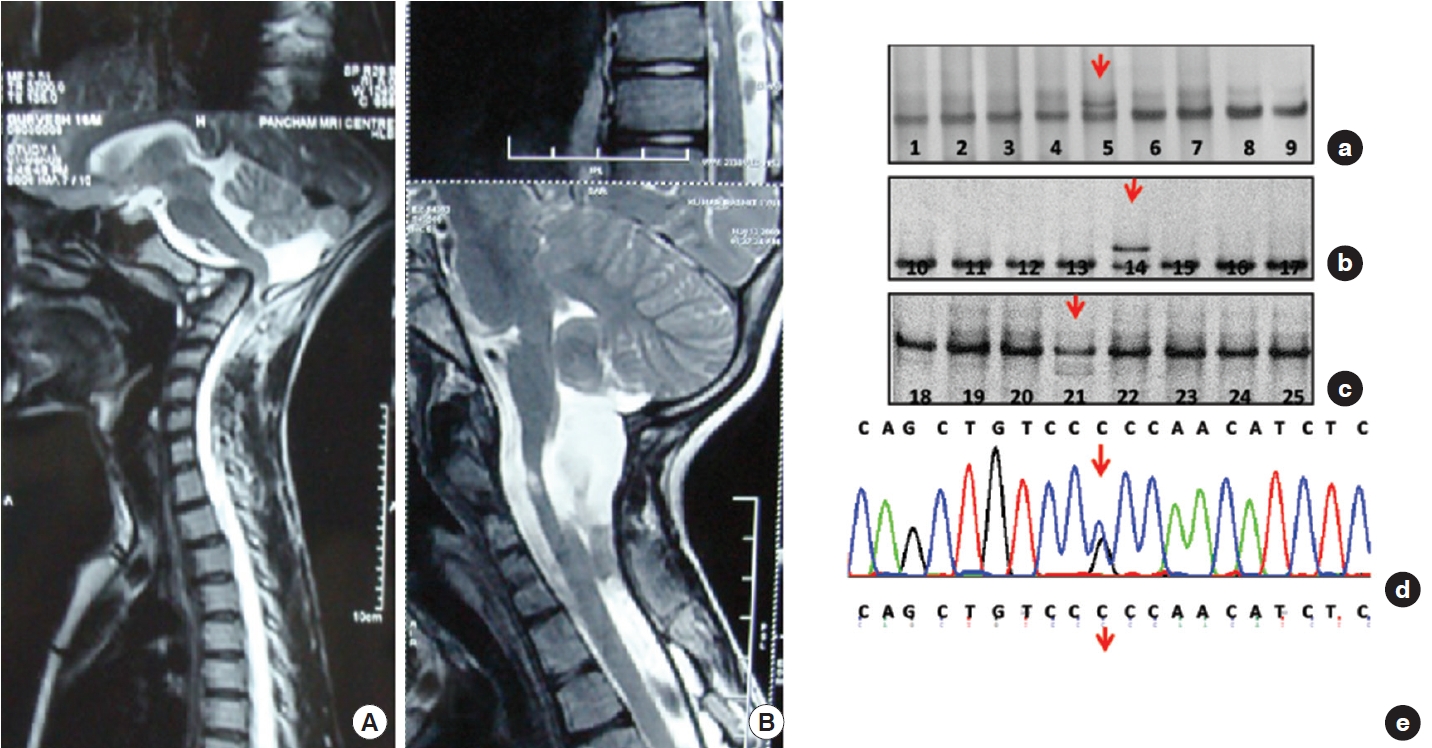
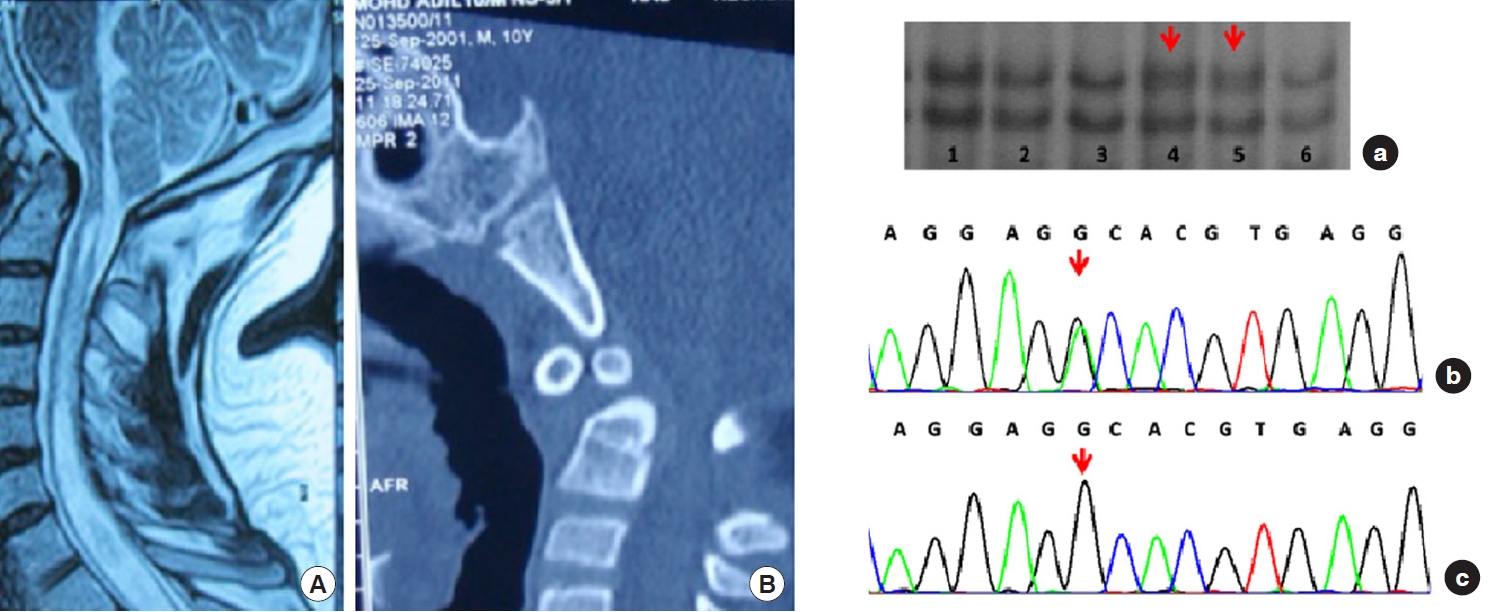
2. Genetic Profile (Figs. 4-7)
Thirty-nine samples (78%) showed sequence variants. Exon 25, 26, 27, and 28 showed variable patterns of DNA bands in SSCP, which on sequencing gives various types of DNA sequence variations in intronic region of the FBN1 gene in 14%, 14%, 6%, and 44% respectively. We observed a total of 7 shifts in 7 patients (14%) in exons 25 and 26 (on sequencing identified as 6 base pair deletion as delTCTTTA). In exon 27, 3 similar shifts were observed (n = 3, 6%). The sequencing here showed a substitution (c.3575C > G), leading to a change in amino acid sequence proline to alanine (P.P1148A). In contrast, exon 28 revealed several shifts in SSCP in 44% of the cohort (n = 22). The sequencing here showed 2 different deletions and one substitution in the intronic region. Other changes include detection of delGTTAT (5-base pairs: del GTTAT, 3589+ 67_3589+71) in 14% (n = 7); detection of delTTTTA (also a 5-base pairs, present in combination with a substitution 3792-5 G/A) in 14% (n = 7). Glutamine/arginine (G/A) was found in with delTTTTA in exon 28. It is also remarkable that the exon 27 (P1148A) change is entirely concordant with the substitution G/A. DNA shifts were not seen in the SSCP gels in the rest of the exons. No shifts were obtained in the control population on SSCP gels in exon 24, 25, 26, and 27, whereas in exon 28, shifts were obtained and identified as delGTTAT. A statistical comparison of genetic differences with the control group could not be done as all the mutations were observed in 78% of the disease cohort and 0% in the control group.
(1) Radiological examination
The study group was comprised of 100 patients (64 males, 26 females) and compared with an equal number of age- and sexmatched controls (total 140 subjects). The mean age of the patients and controls was 21.9±8.2 years.
The values for all the various parameters (patients and controls) are as shown in Table 1. Using receiver operating characteristic curves, the Chamberlain line’s distance, with zero as the cutoff, had the best sensitivity and specificity.
The most frequent anomaly was BI (n = 35, 72%) followed by AAD (n = 33, 66%) and clival segmentation defects (n = 18, 36%). This was followed by CMI (n = 9, 18%), occipitalized C1 (n = 8, 16%), syringomyelia (n = 8, 16%), os odontoideum (n = 4, 8%), platybasia (n = 3, 6%), Klippel Fiel anomaly (n = 2, 4%) and atlas assimilation (n = 2, 4%)
The mean ADI in patients (Table 1) with BI and AAD was 5.5±3.2 mm (the ADI in controls was 1.0±0.3 mm). The mean distance of the dens in patients with BI and AAD above Wackenheim’s clival canal line was 5.5±4.4 mm (in controls, the dens was 2.6±1.8 mm below this line), the mean distance above Chamberlain’s line in patients was 8.3±4.2 mm (in controls, the dens was 2.9±3.3 mm below this line). The mean distance above McRae’s line in patients was 2.5±3.4 mm (in controls, the dens was 5.2±1.2 mm below this line). The mean values of the modified Ranawat’s line were 18.8±4.4 mm and 26.1±2.5 mm for patients and controls. The differences between patients and controls for all values were significant (p < 0.01). Table 2 shows the strength of the atlanto dental interval and various joint indices.
The comparison of the severity of AAD with the various joint indices revealed that there was a significant correlation with the sagittal inclination (SI) and the craniocervical tilt (both p < 0.01) (Table 1). When the severity of BI (Chamberlain’s line) was compared (Tables 1, 2) with various joint indices, it strongly correlated with the SI and craniocervical tilt (both p < 0.01). Besides, the joint reciprocity (p = 0.04 for left side, 0.01 for right side) correlated with the BI. Thus, the joints, which had a higher coronal joint inclination and were nonreciprocal joints, were more prone to develop BI. The correlation with coronal joint inclination (p = 0.16 for left side, 0.2 for right side), joint surface area and overlap index failed to reach significance with both AAD and BI.
The mean SI for controls was 83.35°±8.65°, and in patients with BI and AAD, it was 129°±24.05° (p < 0.01 for both BI and AAD). The mean craniocervical tilt for controls was 60.2°±9.2°, and in patients with BI and AAD, it was 86.0°±18.1° (p < 0.01 for both BI and AAD). The mean coronal joint inclination for controls was 110.3°±4.23°, and in patients with BI and AAD, it was 125.15°±16.4° (the mean p-value between the right and left joints was 0.2, for BI but not AAD). Finally, joint nonreciprocity also correlated significantly with BI and AAD, i.e., the higher the nonreciprocity, the more severe the BI and AAD.
DISCUSSION
In the present study, firstly, we have obtained DNA sequence variants in 39 patients (78%). Secondly, we found a new combination of DNA sequence variants, which were reported earlier in isolation as polymorphism. Still, to the best of our knowledge, no earlier study has reported this combination of sequence variants. Third, the study provides a clue that the “nonsyndromic” bony CV junction anomalies commonly seen in our set up maybe a “softer variant” of MFS. Despite the less known etiopathology, somatic markers such as the short neck, low hairline, low set ears, and systemic anomalies suggest an underlying genetic basis. However, no constant cluster of phenotypic anomalies or spine structural defects has been reported except BI and AAD, which forms the common denominator [45]. The present study’s findings are important from a treatment point of view as it strengthens the fact that one of the essential components of the pathology is the presence of defective ‘holding’ ligaments that have led to progressive telescoping of the cervical spine into the skull base. This provides a fundamental basis for treating the pathology by utilizing joint distraction and deformity correction through a posterior approach rather than performing a trans-oral decompression of the odontoid process [25,46,47]. The latter surgical assumption is based on a hypothesis that this deformity is “irreducible,” a fact that is slowly changing to understand that most of these anomalies are reducible [25,46,47]. Our study provides molecular rationale to this changing concept.
Defects in FBN1 expression results in abnormal elastic fibres, leading to skeletal and cardiovascular anomalies in MFS [48]. The genetic abnormalities in MFS produce a wide range of results, ranging from 10%–100% mutant fibrillin. This results in a substantial phenotypic range of severity [48]. Further, these anomalies are also associated with various other genetic syndromes. Hence, there is a possibility these anomalies of the cervical spine are the variable expression of a connective tissue disorder or a new syndrome in which there is an involvement of connective tissue malformation. Many known genetic disorders that segregate with CM1 affect mesodermally derived cartilage and/or bone [8,49].
In this study, the sequence variants obtained in exon 25 and 26 were identified as a 6 base pair deletion delTCTTTA. It was reported as polymorphism in patients with MFS37and found to be clinically insignificant. The reason behind the presence of this change in both exons is the common intronic region shared by these 2 adjacent exons. Further, in exon 27, 3 patients showed 3 similar kinds of shifts. When sequenced, we obtained a substitution of C > G change (c.3575C > G) in the exonic region leading to a change in the amino acid sequence proline to alanine (P. Pro1148Ala). Pro1148Ala substitution codes for the 13thcbEGF-like domain in the FBN1 gene. Although Pro1148 is not directly involved in calcium-binding, it is conserved in the mouse and bovine FBN1 gene. Proline to alanine changes has also been described in other proteins, leading to various consequences ranging from incorrect folding [50] to a native structure virtually identical to the wild type [51]. Presence of such polymorphism leading to laxity of ligaments may have favoured a posterior approach of joint distraction which has emerged over the past decade as a treatment modality [21,24,25,52]. In a study by Schrijver et al. [53], a homology search for the FBN1 amino acids was performed and suggested that alanine (in the place of proline) does not change the native domain structure or its function. Since previously, the Pro1148Ala substitution had been identified in individuals affected with MFS, aortic disease, and Shprintzen–Goldberg syndrome and numerous unaffected family members, but only once in 367 unaffected control samples [54]. It was considered to be a potential predisposing allele for aortic aneurysm [54]. Substitution of a different proline by alanine in the first EGF-like clotting factor IX domain has been identified in a patient with mild haemophilia B [55]. To explain this substitution’s functional consequences, the authors postulated that altered secondary structure could lead to distortion of cofactor binding sites. In other proteins, proline-alanine interchanges have also been associated with abnormal phenotypes [56,57]. The possibility has to be considered that this substitution may not be wholly neutral but could convey a risk factor in aortic disease. Detailed phenotype-genotype correlations may reveal a modifying influence of the P1148A substitution present in cis or trans. Interestingly, there was complete concordance between the presence of the P1148A substitution and the less common G/A substitution at the intronic region of exon 28. We also obtained complete concordance of this association in 3 patients. Nevertheless, the association is an attractive hypothesis because theP1148A substitution in exon 27 is less than 300 bp upstream of the intron G/A polymorphism [58]. These 2 sequence variants' concordances suggest that this association may be of ancient origin in human evolution. Schrijver et al. [53] also found that P1148A was associated with G/A polymorphism. They suggested that this concordance may be due to linkage disequilibrium.
The new combination of DNA sequence variants that we have found in our study is 5-base pair deletion delTTTTA and a substitution G/A in 11 subjects in exon 28. Both changes were reported in isolation [58,59], but not in combination. More investigation is needed, but there are equal possibilities that this combination may have some indirect role with the “main etiogenic agent” for CVJ anomalies along with evolutionary linkage disequilibrium.
Most of the DNA sequence variants which we have detected in our study were also obtained by Chowdhury et al. [60] in the patients with tetralogy of Fallot (which shows similar histopathologic changes to bicuspid aortic valvular disease, other fibrillinopathies and FBN1 implication in MFS) when screened FBN1 gene (exon 24–28) in Indian patients Exon 25 and 26 showed deletion of TCTTTA. Similarly, in exon 27, they found P1148A substitution in 5 patients. Further, in exon 28, they found 2 types of intronic changes, a 6 base pair deletion (delATTTTT) and a 5-base pair deletion (delTTATG), which we did not obtain. They suggested an increased incidence of “DNA sequence variants” of the FBN1 gene (exon 24–28) may account for or coexist with the higher incidence of aortic dilation in the patients with tetralogy of Fallot. The third change that we have found in exon 28, was the deletion of 5-base pair (delGTTAT) detected in 5 patients and 2 controls, is reported as polymorphism (rs72132658).
The elucidation of the genetic contribution to the developmental CVJ anomaly will undoubtedly aid in diagnostic evaluation and surgical planning, and it will allow more accurate genetic counselling regarding the risk of recurrence to relatives in the immediate future. Even though we did not find any systemic anomalies in our study, an underlying knowledge that FBN1 gene anomalies seen in developmental CVJ anomalies could also precipitate cardiac anomalies should prompt the treating physician more rigorously screen these group of patients. Genetic mechanisms of AAD have not previously been shown. Some of the previous case reports of familial Chiari type I malformation suggests genetic causes. It has been reported earlier that estimates of genetic cases vary from 1/18,000 to 1/1,280. It has been shown that in cases twins with CMI and one set of monozygotic (MZ) triplets, higher concordance observed in MZ twins compared to dizygotic twins. In a separate study involving 364 patients, 12% had at least one close relative with CMI and/or syringomyelia. As CMI is associated with a variety of other genetic conditions, that also provides an evidence for a genetic component. A study showed evidence for linkage to regions on Chromosomes 9 and 15, containing the FBN1 gene, further supports a genetic role in Chiari malformation. In another study, polymorphism has been reported in MTHFR gene, known to have a role in neural tube defects, in patients with AAD. Characterizing the phenotypic presentation and genetic contribution to CMI will not only provide valuable insight into the complex etiology and pathogenesis of the disorder but also aid in associating the risk of symptoms and risk of CMI in relatives of affected patients.
The other component of this study included studying the joint morphology. Our previous studies [20,22-24,26], and the current study reiterated that in developmental CVJ with AAD and BI is associated with increased obliquity of the joints. The severity is directly proportional to the severity of the AAD and BI. This is especially so for values of craniocervical tilt and SI. Besides, presence of increased “obliquity” of joints (increasing angulation of SI:SI) has been the fundamental basis of developing the modifications of the surgical technique of DCER (distraction, compression, extension, and reduction) [19,21,25,26]. The improvements include “joint remodelling” and “extra-articular distraction”. Joint remodelling is performed when the SI is between 90°–110°, and extra-articular distraction is performed when the joints are entirely vertical (SI: 180°). However, it is also to be remembered that the ligaments around the facet joint and those stabilizing the CVJ are quite strong (namely apical, alar, transverse, accessory, and anterior longitudinal ligaments, tectorial, anterior and posterior atlantooccipital membranes). Hence, we hypothesized that only increased obliquity of joints alone might not be responsible for producing AAD and BI. We have also noticed that most patients with developmental CVJ have somatic markers suggestive of an underlying genetic predisposition but do not fit into any specific genetic syndrome (in our cases, clinodactyly, low set ears and increased first interdigital space; Figs. 1, 2). A majority of our patients also have hyperlaxity of joints. Given all the above findings, we decided to study the FB1 gene, which is also affected in MFS. Based on our radiological findings, we propose a new hypothesis for developing AAD and BI in developmental CVJ. We propose that the presence of structural defects in ligaments (due to FB1 gene polymorphisms) and increased obliquity of joints lead to precipitation of AAD and BI. Since most patients develop symptoms by the 1st–2nd decade, we feel that weight-bearing and mechanical stress are further responsible for precipitating instability.
The study has its limitations, like the relatively small sample size. Radiological examination in first degree relatives could not be performed due to either unavailability of the family members or refusal to consent. Another limitation of the study is the variable sensitivity of SSCP.
We also do agree that the exact cause of joint inclination is not known. The actual etio-pathogenesis may be more complex. We wanted to approach the problem in a manner that it could be understood from a clinical perspective. The underlying question was that if the joint morphometry was abnormal, why do most of our patients present between 1st–3rd decade? Effect of gravity obviously plays a role. But the question was again that why do these anomalies present in mostly in younger ages? That is when we thought of exploring the FBN1 mutations which is responsible for encoding for the formation of ligamentous structures and is abnormal in MFS. Additionally, many of our patients presented with phenotypic features of Marfan’s (e.g., hyper-extendibility of fingers etc.). This further prompted us to study this gene. However, further studies will be required in this direction to have more clarity.
CONCLUSION
A significant number of DNA sequence variants in patients with developmental CVJ anomalies signifies that these variants may not be directly pathogenic, but they are showing association genetically. The DNA sequence variants in these anomalies were also reported in MFS. Our study suggests that the nonsyndromic developmental CVJ anomalies so commonly encountered in our clinical situation may be a “subtler version” of MFS or similar connective tissue disorders in which FBN1 is involved. Furthermore, the presence of abnormalities in joint morphology in the form of increased obliquity may also lead to AAD and BI precipitation. The present study is interesting because it provides further guidance to establish a genetic basis for developmental CVJ anomalies. However, more intensive investigations with some more genes along with FBN1 are needed in a large cohort of patients. However, a combination of factors like weakened “holding ligaments,” increased obliquity of joints, weight-bearing, etc., may precipitate the AAD and BI in patients with developmental CVJ anomalies.