INTRODUCTION
Low back pain (LBP) is highly prevalent as age expectancy increases, and it is the leading cause of years lived with disabilities worldwide, affecting at least 600 million people globally [1]. A widely recognized contributor to chronic LBP is the degeneration of intervertebral disc (IVD). The discs are fibrocartilaginous tissues that lie between the vertebrae of the spinal column and provide flexibility to the spine and absorbs the mechanicals loads during diurnal events [2]. During aging, the mechanical properties of the disc are often compromised due to structural changes in the tissue. Disc and cartilage function depends significantly on the integrity and composition of the extracellular matrix (ECM) which consists of intricate and finely organized networks of collagens and proteoglycans (PGs) [3]. PGs through their sulfated glycosaminoglycans (GAGs) provide an osmotic mechanism to attract water molecules into the tissue essential to accommodate compressive and tensile loads on the spine and joint tissues [4]. Aging or genetic mutations in PG genes result in reduced levels or abnormalities which impair their biological functions. This review will focus on major PGs in the IVD and cartilaginous tissue and their contribution to disc pathologies and skeletal dysplasias.
PGs generally consist of a core protein covalently attached with one or more GAG chains joined through a tetrasaccharide bridge at a serine residue. These GAGs, typically long polysaccharides with repeating disaccharide structures, are categorized into 4 groups: chondroitin sulfate/dermatan sulfate (CS/DS), heparan sulfate (HS), keratan sulfate (KS), and hyaluronic acid (HA) [5]. The biosynthesis process of GAGs, specifically HS and CS, is tightly regulated in order to maintain their constant concentration within the tissue. Alteration in HS or CS GAG levels can affect tissue development, physiological, or pathological processes [6,7]. Briefly, the process of GAG synthesis requires 5 uridine diphosphate (UDP) derived activated sugars such as UDP-glucuronic acid (GlcA), UDP-N-acetylglucosamine (GlcNAc), UDP-xylose (Xyl), UDP-galactose (Gal), and UDP-N-acetylgalactosamine (GalNAc) [5]. These sugars are transported from the cytoplasm to the Golgi apparatus where a series of glycosyltransferases assemble the tetrasaccharide bridge. GAG synthesis begins when xylosyltransferases (encoded by XT1 or 2) add Xyl to a serine residue on the core protein, followed by galactosyltransferases I (B4GALT7) and II (B3GALT6) which add 2 Gal sugars. Next, a glucuronyltransferase (B3GAT3) adds GlcA to assemble a common tetrasaccharide bridge containing GlcAβ1-3Galβ1-3Galβ1-4Xylβ1-O-Ser [5,8,9]. The subsequent addition of GalNAc by GALNACT2 or GlcNAc by EXTL3 will then initiate the commitment for either CS or HS biosynthesis [8]. The regulation of GAG biosynthesis in the context of disc biology is further discussed by Silagi et al. and others [9,10]. All GAGs, except HA, undergo a sulfation process in the Golgi where sulfotransferases catalyze sulfate donor compound 3´-phosphoadenosine-5´-phosphosulfate (PAPS) to modulate their sulfation profile [5]. Alterations in sulfation profiles can modulate the critical physiological functions of PGs in developing tissues often leading to chondrodystrophies [11]. Based on the recent classification of genetic skeletal disorders, mutations of ion transporter-related or sulfation-related genes such as SLC26A2, PAPSS2, IMPAD1, CHST3, SLC35B2, CHST14, DSE, CHST11, HS2ST1, SLC13A1 result in achondrogenesis, chondrodysplasia, Ehlers-Danlos syndrome, osteochondrodysplasia, brachydactyly, overlapping malformed digits and developmental delays, and disc degeneration [11-13]. Importantly, defects in matrix recycling by autophagy observed in lysosomal storage disorders are equally detrimental to skeletal tissues and during development [14].
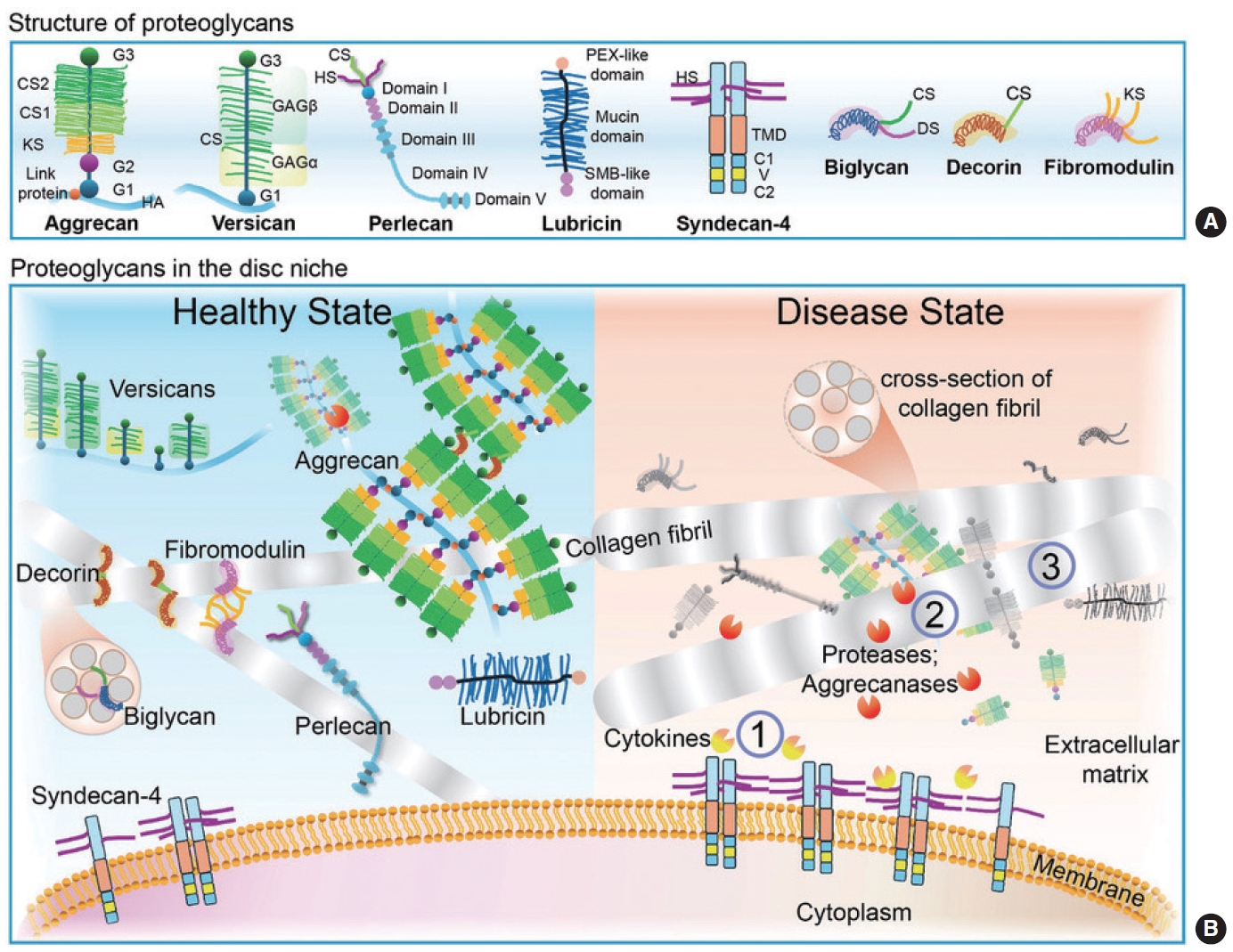
It is important to mention here that PGs were previously classified either by GAG type or PG size due to their heterogeneity. To simplify PG classification, Iozzo and Schaefer proposed to categorize mammalian PGs into 4 overarching classes based on their locations: extracellular, pericellular, cell surface, and intracellular [15]. In the context of the joint, PGs are present on the cell surface and within the ECM of the growth plate, articular cartilages, and IVD. During development, these macromolecules can interact with constituent growth factors, cytokines, morphogens, and chemokines to influence cell adhesion, morphology, proliferation, migration, and differentiation [16]. Major PGs, including aggrecan, versican, perlecan, and cell surface PGs such as glypicans and syndecans, particularly syndecan-4, and a few small leucine-rich proteoglycans (SLRP), such as biglycan and decorin, fibromodulin, and lubricin are abundant in the articular cartilage and the disc to aid in tissue hydration, biomechanical function and cell signaling events (Fig. 1A and B). These will be discussed in detail below (Table 1).
STRUCTURE AND FUNCTION OF PROTEOGLYCANS IN MUSCULOSKELETAL PATHOLOGIES
1. Extracellular PGs
ACAN encodes for aggrecan, the most abundant PG in the IVD and joint cartilage. Aggrecan bears negatively charged KS and CS GAG side chains providing cartilage with its ability to bind water for hydration and withstand large compressive loads [17]. Structurally, aggrecan consists of a core protein with 3 disulfide-linked globular regions (G1, G2, G3) with intervening extended regions between G2 and G3 [18] (Fig. 1A). Between the amino terminal G1 and G2 domains is the interglobular domain (IGD), a prominent proteolysis site that is thought to be involved in the physiological turnover of aggrecan [19]. Following the G2 domain, the core protein is decorated with approximately 30 KS chains and 100 CS chains in the CS1 and CS2 domains; these domains are responsible for the water-binding property and its function as a structural PG. The G3 domain of aggrecan resides at the carboxy-terminal and is a complex region that is required for post-transcriptional processing [19]. Aggrecan does not exist in isolation within the ECM but is instead composed of aggrecan supramolecular aggregates (Fig. 1B). These supramolecular aggregates form when multiple aggrecan molecules noncovalently attach to a link protein-bound HA filament [20]. Molecularly, the G3 domain is essential for sufficient modification of GAGs; without this, the CS-containing constructs are not secreted. In the developing skeleton, aggrecan expression is confined to chondrocytes and other cartilaginous tissues [21].
The importance of aggrecan in cartilage was first illustrated in the embryonically lethal nanomelia affecting cartilage development in chickens [22] and cartilage matrix deficiency (cmd) in mice resulting in cleft palate, dwarfism, abdominal compression, and respiratory failure after birth [23-25]. Interestingly, heterozygous cmd mice appeared normal at birth, however, age-associated skeletal defects such as dwarfism and spinal misalignment began to show at 19-month of age [26]. The underlying mechanism in lethal nanomelia avian mutation is a transversion creating a premature stop codon, truncating part of CS2 and G3 domains and significantly reducing the steady-state level of aggrecan messenger RNA (mRNA) [27]. The residual mRNA gets translated but is not secreted into the ECM [28], thus accumulating in the endoplasmic reticulum [29]. Similar to nanomelia, cmd in mice also results in reduction of aggrecan mRNA expression and secretion of aggrecan into the ECM [26]. These detrimental effects foreshadowed the outcome of ACAN mutation in humans, discussed in a later section.
Versican, another large extracellular proteoglycan encoded by VCAN, is structurally related to aggrecan, possessing a terminal domain analogous to aggrecan’s G1 and G3 regions, however it does not contain IGD, a G2 domain, and KS GAG attachment sites (Fig. 1A) [15]. To date, 5 isoforms of versican (V0–4) have been identified: the full-length versican (V0) and 3 splice variants that lack GAGα (V1), GAGβ (V2), both GAGα and GAGβ (V3), and portion of GAGβ (V4) [15]. During development, full-length versican is prominently expressed throughout the IVD [30]. As the disc matures, its expression decreases throughout but remains prominent between the lamellae of the annulus fibrosus (AF) [30,31]. Functionally, based on its structure and localization with elastin and fibulin-1, a glycoprotein that is incorporated into fibrillar ECM, versican is suggested to contribute to the organization of the disc ECM and provide structural support and resilience to mechanical forces [31]. In cartilage development, the balance of versican expression is important in mediating local transforming growth factor beta (TGF-β) in chondrocyte differentiation and digit joint formation [32-34]. Reductions in versican can compromise chondrogenesis and synovial joint development [35]. Mutations in the human versican gene result in the dominantly inherited Wagner syndrome, caused by a base substitution mutation of VCAN at exon 8 that produces less V1 and more V2 and V3 isoforms, which is associated with progressive vision loss [36].
Encoded by PRG4, proteoglycan 4 or lubricin is a mucinous glycoprotein and an atypical proteoglycan that covers the cartilage and prevents it from cartilage damage. Its structure consists of 2 adhesive nonglycosylated subdomains flanking a heavily glycosylated and mucin-like central domain region. The amino terminus possesses a somatomedin-B-like domain while the carboxyl terminus has a hemopexin domain, both of which are responsible for mediation of cell-cell and cell-matrix interactions to promote cell attachment [37,38] (Fig. 1A). The central domain mostly consists of GalNac, Gal, and NeuAc sugar groups O-linked at threonine residue, making it negatively charged and creating a strong repulsion via hydration forces [37]. Lubricin is found in the synovial joint, coated on the cartilage surface, and detected at markedly higher levels in the IVD compartments compared to other cartilage subtypes [39,40]. Lubricin deletion (Prg4-/-) in mice by excision of exon 6, removing mucin-like domain, results in age-associated accelerated synovial hyperplasia, abnormal protein deposits on the cartilage surface, and disappearance of underlying superficial zone chondrocytes ultimately contributing to joint failure [41]. Since lubricin is detected at a high level in the disc, its absence may be consequential to its integrity as discussed later.
Biglycan is a small ECM PG that is ubiquitously expressed in the articular regions, epiphyseal cartilage, vascular canals, and periosteum during development [42]. It was reported to contribute to bone growth, muscle development and regeneration, and collagen fibril assembly. Biglycan contains 2 CS or DS GAG chains at serine-glycine attachments sites in the N-terminal region (Fig. 1A). It can bind to TGF-β [43], and modulate bone morphogenetic protein (BMP)-4 to influence osteoblast differentiation and maturation in bone development [44]. Located on the X chromosome region Xq28, genetic mutation of BGN affects males more than females. Early work on BGN deficient male mice (BGN-/0) showed early onset of osteoporosis-like phenotype, skeletal abnormalities, and disc degeneration [45-47]. Closely related to biglycan is the SLRP decorin, sharing > 65% of its overall homology. It is encoded by DCN and only has one GAG chain (Fig. 1A). It is known for decorating along fibrillar collagen and regulates the association of collagen into proper fibrils protecting them from proteolysis [15] (Fig. 1B). Deletion of decorin in mice (Dcn-/-) results in fragile skin and tendons along with coarser and irregular fiber outlines without affecting bone mass [48]. In cartilage, decorin mediates matrix integrity and biomechanical functions by enhancing the linkages and assembly of aggrecan-aggrecan molecules and aggrecan-collagen II fibrils [49]. Since biglycan and decorin are highly homologous and co-expressed in tissues such as skin and bone, double deletion of biglycan and decorin (BGN-/0 Dcn-/-) mice revealed compounding effects on skin fragility, severe osteopenia, and alteration of collagen fibril structure and mechanical properties of tendons [50,51]. SLRPs fibromodulin and tenomodulin are also present in cartilage, tendon, and ligament ECM and contribute to the structural integrity of these tissues. Fibromodulin, encoded by FMOD, is a KSPG SLRP highly homologous with biglycan and decorin (Fig. 1A). It bears KS chains and binds to collagen I via residues located in leucine-rich region 11 in the convex surface of the protein core at a different region than decorin binding site [52,53]. Specifically, it interacts with triple-helical type I and II collagens [54], regulates fibrillogenesis and collagen fibril size [55], and maintains long-term tissue integrity within the knee [56] (Fig. 1B). During collagen proteolysis, fibromodulin gets cleaved by a disintegrin and metalloproteinase with thrombospondin motifs 4 and 5 (ADAMTS-4 and-5) and matrix metalloproteinase-13 (MMP-13) [57,58]. Deletion of fibromodulin in mice (Fmod-/-) results in abnormal tissue organization in the cross-sections of the tail, a larger proportion of thin collagen fibril diameter in the Achilles tendons, higher histological arthritis score, and abnormal dentin mineralization and alveolar bone formation [55,56,59-61]. Similar to BGN-/0 Dcn-/- double knockout, biglycan and fibromodulin double knockout (BGN-/0 Fmod-/-) mice have impaired collagen fibrils in the tendons. This leads to gait impairment, increased ectopic tendon ossification due to increased use of leg joints, and severe premature osteoarthritis [59]. Furthermore, these mice develop accelerated temporomandibular osteoarthritis due to accelerated chondrogenesis secondary to reduced levels of sequestered TGF-β1 in the ECM, leading to overcompensation of overactive TGF-β1 signal transduction [62]. Tenomodulin regulates tenocyte proliferation in tendons [63] and is required for proper collagen I cross-linking; deletion of tenomodulin (Tnmd-/-) in mice resulted in abnormal collagen I cross-linking and increased collagen fiber thickness and stiffness, leading to inferior endurance running performances [64,65].
2. Cell Surface PGs
Syndecans have a single-pass transmembrane protein core which includes an ectodomain bearing HS-chain or HS/CS chains, a transmembrane region, and an intracellular cytoplasmic domain. The cytoplasmic domain comprises of 2 conserved constant regions, C1 and C2, flanking a variable region, V1, that are responsible for syndecan-specific signaling [66-69] (Fig. 1A). There are 4 syndecan family members: syndecan 1-4 (encoded by SDC1-4). Sdc1 and Sdc3 are the largest family members bearing 2 CS chains and several HS chains, while Sdc2 and Sdc4 are smaller and only bear HS chains [67]. All are intrinsically disordered and dynamic, enabling them to interact with numerous ligands including fibroblast growth factor (FGF), vascular endothelial growth factor (VEGF), platelet-derived growth factor, BMP-2, and Indian hedgehog (IHH) [66,70]. While Sdc1-3 are found in specific tissue types, Sdc4 is ubiquitously expressed in most tissue types. Sdc4 functions as a receptor or co-receptor to ligands strengthening the duration and intensity of downstream signaling. It can also form physical connections with the ECM and activate mechanosensory signaling to influence cytoskeletal reorganization during migration and the assembly and disassembly of integrin complexes at focal adhesion sites [66,71]. During early embryo development of Xenopus laevis, gain- and loss-of-function experiments showed that the balance of Sdc4 expression was crucial in regulating the convergent-extension movement in neural tube closure and neural crest-directed migration through the noncanonical Wnt pathway by its interaction with Frizzled 7 (Fz7) and Dishevelled (Dsh) [72]. Similar to X. laevis, Sdc4 is expressed during murine development in the cranial neural folds on embryonic day 8–8.5 and is regulated by Vangl2 during neural tube closure [73]. Unlike X. laevis, Sdc4-null mice have no obvious developmental defects besides delayed wound healing, impaired angiogenesis, and defects in muscle regeneration after damage and myogenic satellite cell differentiation [74-78]. In recent years, we and others have reported that cytokines tumor necrosis factor alpha (TNF-α) and interleukin 1 beta (IL-1β) induce Sdc4 expression to mediate different musculoskeletal pathologies including rheumatoid arthritis, osteoarthritis (OA), and IVD degeneration [79-81]. Moreover, inhibition or blocking of Sdc4 under inflammatory conditions prevents exacerbation of cartilage deterioration [79].
3. Pericellular Proteoglycan
HSPG2 encodes for the secreted perlecan, a large basement membrane HS proteoglycan presents in several ECM tissues including cartilage and the disc. The core protein of perlecan consists of 5 domains (domain I–V) with tandemly repeating modular motifs resembling “pearls-on-a-string,” 2 of which (domain I and V) possess HS GAG attachment sites [82] (Fig. 1A). Perlecan expression has been well characterized in the early stages of embryogenesis in murine models. It appears as early as E10.5 in the heart, pericardium, blood vessels, and developing vertebral cartilage [83] and is highly expressed in cartilage primordia at E15.5 [84]. Perlecan is expressed ubiquitously in muscle and bone marrow enabling homeostatic regulation of biological processes, such as the formation of cardiovascular tissue, localization of acetylcholine esterase to neuromuscular junctions [85], and formation of bone [86]. Ablation of perlecan in mice (HSPG2-/-) resulted in significant embryonic lethality due to pericardial hemorrhaging from defects in basement membranes and biomechanical deterioration of the contracting myocardium between E10–12 [87,88]. However, the few mice that survived birth died with skeletal dysplasia characterized by micromelia, narrow thorax, and craniofacial abnormalities, similar to skeletal defects of patients with lethal dyssegmental dysplasia, Silver-Handmaker type (DDSH) [88]. Furthermore, perlecan-null cartilage exhibited reduced GAGs, disorganized collagen fibrils, diminished chondrocyte proliferation, and prehypertrophic zones; all of which contributed to abnormal skeletal phenotypes similar to those of patients with thanatophoric dysplasia type I, which is caused by activating mutations in FGFR3 [88]. Since no homozygous knockout mice survived, hypomorphic HSPG2C1532Y−Neo mice were generated to study nonlethal skeletal dysplasia mimicking Schwartz-Jampel syndrome (SJS) in humans [89]. These hypomorphic HSPG2C1532Y−Neo mice have C1532Yneo mutation in domain III of perlecan, resulting in reduced perlecan expression, reduced cellular and ECM stiffness and defective pericellular matrix formation, potentially from impaired incorporation of newly synthesized ECM [89-91]. Given that perlecan was also observed in developing vertebral cartilage, another perlecan murine model lacking exon 3 (HSPG2Δ3/Δ3) was generated to study the role of HSPG2 in the disc. Perlecan exon 3 encodes perlecan domain I, and its deletion in HSPG2 exon 3 null mice resulted in a 22 kDa size reduction in the perlecan core protein [92]. As a result, HSPG2Δ3/Δ3 mice had a higher GAG content in IVDs, advanced chondrocyte hypertrophy in the cartilage, and disorganization of the growth plate [92].
PROTEOGLYCAN-LINKED DISC PATHOLOGIES
The IVD is an avascular organ composed of a collagen-rich AF that encompasses the gelatinous PG-rich nucleus pulposus (NP) center; both compartments are anchored by superior and inferior endplates composed of a hyaline cartilaginous and bony endplate region. In young healthy disc tissue, the NP ECM is mainly composed of aggrecan and versican whose functions are to elevate water swelling potential and resist spinal compression. These PGs bear negatively charged CS side chains that draw in hydrated osmolytes, such as cations (Na+, Ca2+), ultimately creating a hyperosmotic environment [9]. TonEBP/NFAT5 is the only known mammalian osmo-sensitive transcription factor that plays an important role in NP cell osmoadaptation and survival within hyperosmotic environment of the disc [93-95]. AF, on the other hand, is composed of mainly collagens I and II to provide tensile strength. In degenerative disc pathologies under the aging or injury contexts, there is a marked loss of water-binding PGs and an increase of ECM remodeling that results in biochemical and biophysical changes in the disc [4]. These changes include decreased PG synthesis and increased aggrecan proteolysis by ADAMTS-4 and-5 and MMPs [96]. In addition to these proteases, cytokines including TNF-α, IL-1β, and IL-6 are important mediators of degenerative cascade under the inflammatory milieu [97].
Aggrecan is an essential component in the disc. As the disc ages, decreased aggrecan levels and functionality is observed. This is either due to a decrease in PG synthesis or an increase of PG degradation, shifting the balance from a proteoglycan-rich to collagen-rich profibrotic environment. In relatively healthy human NP tissue that correlated with gross morphologic Thompson grade of 2 (1 being the healthy and 5 being the most degenerated state), the GAG to hydroxyproline ratio was about 23:1; however, in the tissue with a Thompson grade of 4, the GAG to hydroxyproline ratio was reduced to about 5:1 [98,99]. As discussed earlier, cmd heterozygous mice cause spinal misalignment and movement problems with age due to impaired secretion or truncation of aggrecan [26]. To characterize the phenotypic and morphologic effects of aggrecan deletion on skeletal development, Lauing et al. [100] developed an ACAN mutation disease model in cmd mice (cmdbc/cmdbc) where the entire protein-coding sequence was deleted. This yielded severe skeletal defects in the limbs, ribcage, and vertebrae development with abnormal mRNA expression patterns of Col10a1, Sox9, Ihh, Ptch1, and Fgfr3 in the growth plate. However, when homozygous cmdbc was rescued with chick aggrecan transgene (cmdbc/cmdbc;Agc/+), skeletal defects were reversed by 20% in limbs and 80%, near-full reversal, in size and diameter of the ribcage and vertebrae.100 This suggests that aggrecan has a major role in regulating key growth factors during development, especially in the development of axial skeletal structures like ribcage and vertebrae. Noteworthy, these detrimental effects likely foreshadowed the outcome of ACAN mutation in humans. While the ACAN gene mutation has not been specifically reported to cause human degenerative disc disease, heterozygous ACAN mutation is associated with both idiopathic short stature and accelerated bone aging which predisposes to early onset of lumbar disc degeneration or herniation [101,102].
As mentioned, loss of aggrecan in disc pathology may be due to increased matrix degradation. Barre et al. [103] reported a significant increase in Sdc4 mRNA expression in damaged human cartilage cultured primary monolayers compared to normal cartilage tissue, suggesting Sdc4 was a likely contributor to the disorganization of cartilage and the development of OA processes. The Risbud group was the first to show enriched Sdc4 expression within the disc tissue [104] and the importance of HS synthesis in regulating matrix catabolism under an inflammatory milieu, mediating ADAMTS-5 activity to cleave aggrecan [81]. Specifically, our findings showed that cytokines TNF-α and IL-1β regulate Sdc4 expression via the nuclear factor kappa B (NF-κB)-p65/RelA dependent mechanism to activate ADAMTS-5 [81] and induce expression of MMP-3 through the mitogen-activated protein kinase–NF-κB axis [105]. Importantly, analysis of degenerated human NP tissues showed a strong correlation between Sdc4, ADAMTS-5, and cleaved aggrecan neoepitopes [81]. To investigate whether Sdc4 deletion could delay the disc degeneration, we characterized the spinal phenotype of Sdc4 null mice. Our findings show that Sdc4 deletion results in early-onset osteopenia and alters biomechanical properties in the lumbar vertebrae due to the dysregulation of osteoclastogenesis. Congruent with this finding, a previous study demonstrated that the GAG-bearing ectodomains of syndecans 1–4 suppressed osteoclast differentiation [106]. Furthermore, discs of adult Sdc4 knockout showed alterations in mature collagen crosslinking, CS content, and aggrecan turnover by aggrecanase. Interestingly, histological assessment of Sdc4 knockout mice showed subtle cellular phenotypes with NP cells containing larger vacuoles and a thicker NP cell band. Importantly, the transcriptomic analysis suggested that Sdc4 deletion downregulates genes associated with mitochondrial metabolism, autophagy, ER to Golgi protein processing, and HS biosynthesis and GAG degradation. These findings indicate that deletion of Sdc4 reduces matrix turnover which is speculated to be responsible for CS accumulation and GAG degradation in NP tissue [107]. Other investigators using Has2- and Sulf1-knockout mice have demonstrated the important of GAG function in PGs. Changes in hyaluronan synthesis and sulfation of GAG profiles can alter the structure and function of PGs. Specifically, HAS2 and SULF1 are important in HA synthesis and sulfation of GAGs, respectively, and their deletion resulted in defects of skeletal development, formation of IVDs [108], and disc degeneration [109].
Collagens are also important structural molecules that provide structural integrity to the disc. Biglycan, tenomodulin, and lubricin are all key mediators in helping collagen maintain its organization, structure, and lubrication. BGN deficient (BGN-/0) mice exhibited loss of notochordal cells at 6 months with advanced degenerative changes at 9 months. These changes were thought to have been caused by a loss of structural stability in collagens, leading to increased mechanical stress resulting in premature disc degeneration [47]. A study of Tnmd-/- mice also showed advanced degenerative changes in the NP and inner AF of the disc, increased hypertrophic-like chondrocytes and apoptosis, decreased disc height index, and smaller collagen fibrils with lower compressive stiffness. In short, decreased proliferation compounded with compromised collagen biomechanical integrity led to defects in the tissue’s ability to twist and withstand mechanical loads which subsequently increased the likelihood of degeneration and disc tears [110]. Lubricin is another important glycoprotein critical in providing joint lubrication and in aiding the endurance to mechanical strain. Lubricin knockout (Prg4-/-) mice were reported to have thinner AF, increased articular surface friction, early progressive surface damage, and increased apparent torsional modulus at L1/L2 disc [111]; all of which highlight the functional importance of lubricin as a protective barrier in the joints.
PROTEOGLYCAN CORE PROTEIN DISORDERS-LINKED SKELETAL DYSPLASIAS
Since PGs are important in conferring mechanical, biochemical, and physical properties to tissues, mutations in genes encoding core proteins can detrimentally affect skeletal tissue development and integrity. Skeletal dysplasias noted in this review are linked to gene mutations in aggrecan (ACAN), biglycan (BGN) and perlecan (HSPG2) [12] (Table 2).
1. Aggrecanopathies in Skeletal Dysplasia
In humans, ACAN mutations result in autosomal-dominant spondyloepiphyseal dysplasia Kimberley type (SEDK) and autosomal-recessive spondylo-epi-metaphyseal dysplasia (SEMD) [112,113]. Autosomal-dominant SEDK is a skeletal dysplasia characterized by a stocky short stature and early-onset progressive joint OA. Anderson et al. first reported about SEDK identified in a multigenerational South African family of UK. white descent [114]. Subsequently, Eyre et al. [115] and Gleghorn et al. [116] performed linkage studies and identified a novel locus on chromosome 15q26.1 where a single base-pair (bp) insertion introduced a frameshift of 212 amino acids that caused a premature stop codon in ACAN, truncating aggrecan protein to lack half of the CS1 domain, the complete CS2 domain, and the G3 domain. Another autosomal-dominant mutation type led to short stature with accelerated bone maturation, early onset of OA, and craniofacial, limb, and vertebral abnormalities [117-120].
The autosomal-recessive SEMD was reported by Tompson et al. [113], describing 3 siblings with SEMD to have had extremely short stature, brachydactyly, distinct severe midface hypoplasia, short necks, barrel chests, lumbar lordosis, and macrocephaly. DNA sequence analysis of affected individuals revealed a missense mutation that predicted an amino acid substitution in the C-type lectin domain within the G3 domain [113], resulting in a reduction of aggrecan secretion [121]. A recent case reported by Fukuhara et al. [122] described an individual with SEMD caused by a heterozygous missense mutation in ACAN, however, the skeletal phenotype noted was much milder than the previous case, suggesting that mutations on different domains of ACAN can lead to different phenotypes.
2. BGN-Linked SEMD
Clinically, BGN mutation leads to X-linked SEMD [123] and Meester-Loeys Syndrome, a connective tissue‐arterial aneurysms disorder [124]. Both disorders are characterized by skeletal dysplasia with short stature. To date, there are only 3 cases of X-linked SEMD (SEMDX) observed in an Italian, Korean, and Indian family [125]. This SEMDX disorder was caused by a missense mutation in BGN on chromosome Xq28 and phenotypically resulted in the shortening of limbs, bowing of the legs, and lumbar lordosis.
3. HSPG2-Linked Skeletal Dysplasia
The generation of perlecan deficient and hypomorphic mice was key to identifying HSPG2 mutations in human autosomal-recessive genetic diseases: DDSH and SJS [88,126,127]. Clinically, DDSH is a rare autosomal-recessive skeletal dysplasia with anisospondyly and micromelia caused by perlecan truncation, resulting in diminished perlecan secretion. The mutation in patients was created by 89-bp duplication of exon 34 of HSPG2 or a frameshift mutation that causes truncation in the perlecan protein core [92,126,128,129]. SJS is also a rare autosomal-recessive disorder characterized by a spectrum of abnormal neuromuscular functions and skeletal dysplasia, such as continuous contractions of muscles throughout the body including the face, abnormal spinal curvature, and shortening of the bone [92,127,130,131]. Patients with SJS survive and exhibit milder phenotypes compared to DDSH patients due to partially functional secreted perlecan [126]. These phenotypes underscore the importance of perlecan in maintaining both cartilage integrity and muscle excitability.
CONCLUDING REMARKS
PGs are essential for proper skeletal development. Within the ECM, large PGs interact with growth factors and osmolytes to confer water-binding properties, tissue hydration, and bioavailability of growth factors, while smaller PGs regulate collagen fibril formation in the ECM. At the cell membrane, PGs can provide stabilization for ligand-receptor interactions, and potentiate signaling complexes that regulate growth factor sensitivity, cell migration, proliferation, and matrix adhesion. Studies have shown that gene mutations of major PGs such as aggrecan, perlecan, lubricin, biglycan, and tenomodulin result in disc degeneration and skeletal defects in murine models. These models described here could potentially be used to explore treatment modalities to restore ECM functionality during disc degeneration. In humans, proteoglycan gene mutations result in severe skeletal dysplasia, including lumbar disc herniation in some cases. Currently, there are no treatments for genetic skeletal disorders except corrective surgical procedures including osteotomy and spinal stenosis surgery.
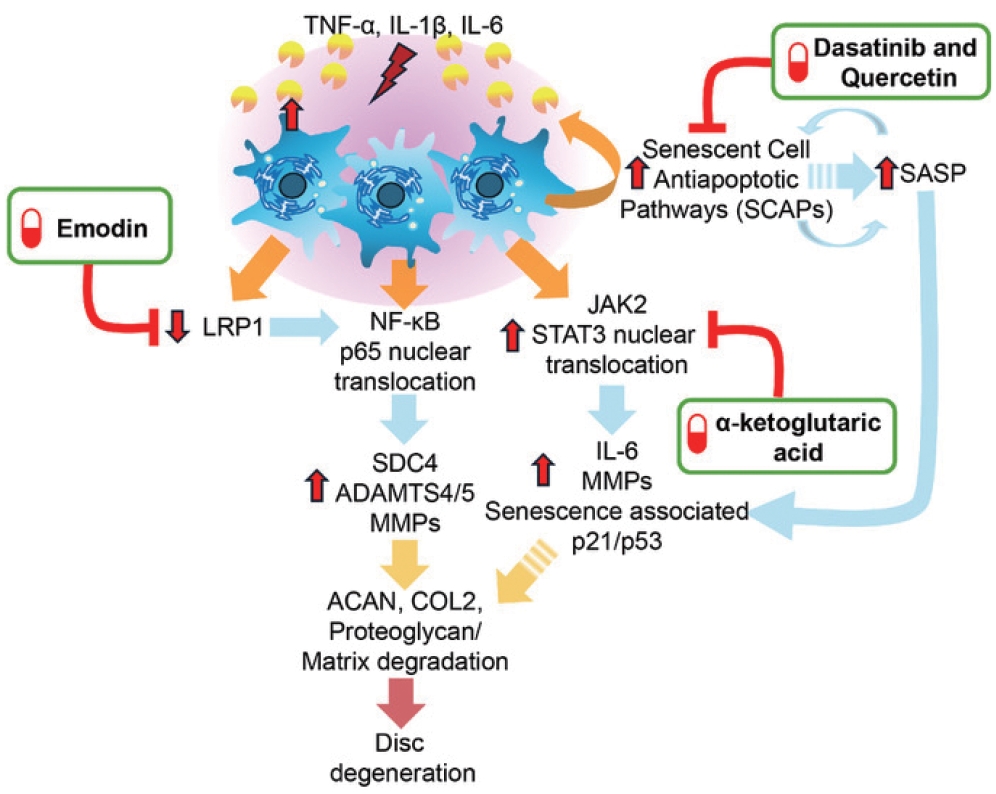
Before we conclude, it is important to discuss a few emerging approaches to treat disc degeneration and restore the matrix function. During aging, senescent NP cells accumulate in the disc [132-134]. Senescent cells are characterized by the senescence-associated secretory phenotype factors, particularly their secretion of proinflammatory ILs and several proteases including MMPs, ADAMTSs and serine protease HTRA1. To minimize age-associated disc degeneration, restore matrix function and to promote the reparative processes, senolytic drug cocktail of Dasatinib and Quercetin (D+Q) and pentosan polysulfate, a HS biomimetic, are under investigation as attractive options [135,136]. The senolytic (D+Q) cocktail works by selectively inducing senescent cell apoptosis by targeting senescent cell antiapoptotic pathways [137,138]. It should be noted that it is technically challenging to deliver biomimetics to inner disc compartments without causing structural alterations during delivery. In contrast, systemic administration of Dasatinib and Quercetin was found to be effective in murine models and is a viable therapeutic option in preventing age-associated disc degeneration and restoring matrix quality. Similarly, long-term systemic administration of a supplement called alpha-ketoglutaric acid was reported to attenuate inflammatory- and age-associated disc degeneration by suppressing catabolic IL-6 expression and preventing JAK2/STAT3 (Janus tyrosine kinase 2/signal transducer and activator of the transcription 3) phosphorylation involved in the degenerative process [139]. In the context of injury-induced disc degeneration, Emodin, a bioactive anthraquinone compound, has been proposed as a potential therapeutic to alleviate inflammation when injected into the injury site [140,141]. This bioactive works by upregulating low-density lipoprotein receptor-related protein 1 to inhibit NF-κB mediated degradation of MMPs and ADAMTS-5, ameliorating disc degeneration and preserving aggrecan expression and functionality in vitro and in vivo [140] (See Fig. 2 for summary of these potential therapeutics). Further clinical trials and controlled studies will be required to assess the efficacy of these therapies.