INTRODUCTION
Chordoma is a tumor arising from embryonic notochordal remnants. It accounts for 17.5% of primary spine tumor, with a reported incidence of 0.5 to 0.8 per 1,000,000 population in the United States8,15). Lesions arise from the sacrococcygeal region (50%), the base of the skull (35%) and vertebral column (15%)4,8,15). Chordoma is classified as a malignant bone tumor, but slow growing feature is distinguished from other malignant bone tumors22,23).
En bloc excision is the treatment of choice for chordoma. However, local recurrence is frequent because complete surgical removal of the tumor is difficult12,13,24). Radiation therapy (RT) is often used as an adjuvant treatment modality and appears to reduce the incidence of local recurrence. However, the effect of RT on overall survival was not known exactly3). Chemotherapy for a chordoma has been proven as an ineffective modality5,17,18).
Dedifferentiated chordoma (DC) is a chordoma with sarcomatous components such as malignant fibrous histiocytoma most commonly, fibrosarcoma, osteosarcoma, or rhabdomyosarcoma rarely21). DC is, for the most part, a transformation type, however de novo type is rarely reported16,21). Transformation type has been reported as a result of malignant transformation of recurrent chordoma or post-irradiation changes12,13,24). The clinical course of DC is distinguished from conventional chordoma by the rapidity of tumor growth and the potential for distant metastasis.
In this article, we report two cases of DC in recurrence.
CASE REPORT
Case 1
A 66 years old male patient visited our hospital with buttock pain. Magnetic resonance (MR) image showed a tumor in the 3rd and 4th sacrum. Percutaneous biopsy was performed and the tumor was diagnosed as chordoma in histopathologic examination. Conventional RT was delivered in total dose of 55Gy. After the RT, the size of the tumor decreased and the buttock pain was relieved.
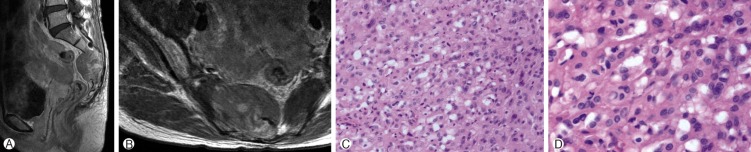
Six years after the RT, the tumor recurred at the original site. Additional RT was conducted in total dose of 32Gy. Ten months after the second RT, the buttock pain was aggravated and MR image showed the increase in the size of the mass, which extends into the 2nd sacrum(Fig. 1A, B). Partial resection below the 3rd sacrum was performed and the tumor was diagnosed as DC in histopathologic examination, which was different from the biopsy result previously reported.
Follow up MR image performed 4 months after surgery revealed the extending tumor into the 1st sacrum, soft tissue around the sacrum and rectum(Fig. 1C, D). Soft tissue mass excision and colostomy was done. Three months later, the tumor penetrated through the operation wound resulting in wound infection. As a result, the patient died of sepsis. There was not any distant metastasis until death. The survival time of the patient after the diagnosis of dedifferentiated chordoma was 12 months.
Case 2
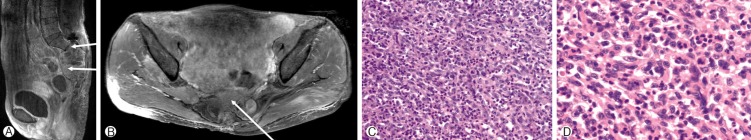
A 41 years old female patient visited our hospital complaining of right buttock pain, perianal hypesthesia and urinary difficulty. MR image demonstrated a solid mass from the 2nd to 5th sacrum extending into presacral soft tissue and sacral spinal canal (Fig. 2A, B). Percutaneous biopsy was performed and the tumor was diagnosed with chordoma (Fig. 2C, D). Gross total resection was done and the buttock pain relieved.
Four months after the operation, buttock pain recurred. MR image revealed the recurrence of the tumor (Fig. 3A, B). The second operation was done. However, gross total removal was impossible because of the tumor invasion into the rectum. The histopathologic result is different from that of the first operation, and the tumor is diagnosed with DC(Fig. 3C, D). The patient did not experience any RT before diagnosed as DC. Conventional RT was delivered in total dose of 66Gy.
Several times of surgical removal and Cyberknife® (Accuray, Inc., Sunnyvale, CA, USA) radiosurgery was done, which could control the localized tumor, however, multiple new lesions developed aggressively and distant metastases to lung and liver occurred. Two cycles of chemotherapy using 4 drug regimen including vincristine, ifosfamide, doxorubicin and etoposide following one cycle of single regimen of etoposide were delivered. However, chemotherapy was not effective to control the systemic tumor. The patient died 31 months after the diagnosis of DC.
DISCUSSION
The incidence of DC has been reported to be 6 to 9% of all the chordomas16,21). DC was characterized as a sharp demarcation of a conventional chordoma with a high grade sarcomatous component16). The histological appearance of sarcomatous component has been described as malignant fibrous histiocytoma, fibrosarcoma, osteosarcoma, or rarely rhabdomyosarcoma21).
The differential diagnosis between DC and conventional chordoma is important because of the distinctions in clinical prognosis16,23). Conventional chordoma is a slow growing tumor with a benign behavior and an indolent clinical course. In contrast, DC is characterized by the potential for distant metastasis and rapid progression. The metastasis commonly contains the dedifferentiated component. Dedifferentiated tumor arising de novo is rarely reported16,21). Sarcomatous change following radiation has been observed more frequently, but a causal relationship is not proved yet10,11,14).
It is difficult to distinguish conventional chordoma from DC based on imaging studies. Typical findings on a plain film of chordoma include osteolytic lesions of the bone with sclerotic bone reaction7). Computed tomography (CT) scan or MR image studies are indicated to evaluated the extent of the tumor and to identify the tissues that the tumor has infiltarated1,19,20). The characteristics of chordoma in CT scan are an expanding, destructive, osteolytic lesion with an associated soft tissue mass. MR image has a better resolution of the soft tissue component than CT scan. Chordoma shows hyperintensity in T2 and hypointensity in T1 weighted images on MR image.
Chou et al.6) reviewed 16 cases of DC previously reported since 1970 in their article published in 2009. Seven out of 16 cases belonged to the de novo type while 9 cases to the transformation type. The median age of patients at the time of diagnosis of DC is 63 years old (range 24 to 73). The male to female ratio is nearly 2:1. The median transformation interval from conventional chordoma to DC in 9 cases was 6.6 years (range 2 to 26). Seven of nine patients experienced local RT before the transformation of chordoma into DC. Nine of the sixteen patients had distal metastatic lesions and lung metastasis presented in all the nine patients. Other metastatic sites include the pleura, inguinal and paraaortic lymph nodes, bone marrow, heart, and spine. The average survival time of all patients was 10.1 months (range 3 weeks to 4 years). De novo group had the average survival of 9.6 months (range 3 weeks to 2 years), transformation group had 10.4 months (range 1 month to 4 years).
In our study, 2 cases were both transformation types, with one male and one female patient. In case 1, the patient's age was 72 years old at the time of the diagnosis of DC, and there was a history of RT. The transformation interval was 6 years, and there was no distant metastasis. The survival was 12 months after the diagnosis of DC. In case 2, the patient was 41 years old at the time of the diagnosis. Interestingly, the patient did not receive any RT before the transformation of the chordoma. In addition, the transformation interval was only 4 months. The survival time was 31 months after the diagnosis of DC. Both of the 2 patients showed rapid disease progression, the second patient lives longer than the first in spite of short transformation time and multiple systemic metastases. Of course multiple factors influenced the survival time of the second patient, an aggressive treatment might be helpful to prolong the survival time.
The treatment of choice for DC seems to be a surgical resection. In Chou et al.6), 6 of the 16 patients had undergone surgical resection alone for DC. A disease free status for more than 5 months was achieved in 3 of the patients. The other 3 patients had recurrent disease postoperatively. Two of the six patients expired at 3 and 8 months postoperatively as a result of the recurrent disease. Four of the sixteen patients had recurrent or metastatic disease, underwent surgical resection followed by local RT to the sacrum. The poor outcomes in these patients may have been due to the difficulty in resection of locally advanced disease, which in turn may have led physicians to boost local RT as adjuvant treatment postoperatively. RT is particularly limited as a primary treatment modality for DC.
In our study, after the diagnosis of DC, there was a difficulty in complete surgical removal of the tumor, so it wasn't achieved. We performed RT and Cyberknife® radiosurgery; however, the patients presented repeated recurrence and distant metastasis. In case 1, even if there were no distant metastasis, local tumor control was not accomplished in spite of several operations and 2 times of RT. In case 2, it seemed like there was a local control for certain duration by postoperative adjuvant RT, multiple recurrence occurred. Chemotherapy also had limited effect.
Felming et al.9) reported the use a 6 drug regimen including etoposide, cisplatin, vincristine, dacarbazine, cyclophosphamide, and doxorubicin for 2 patients with DC who presented with multiple lung metastases. In one patient, complete response was achieved without recurrence for 24 months follow up period. Partial response to the 6 drug regimen was achieved in the other patient. However following salvage therapy with ifosphamide could achieve a complete response. The patient died with recurrent disease 28 months after DC was diagnosed.
Meis et al.16) reported the use of a doxorubicin based regimen in 2 DC patients with lung metastasis. In one patient partial response was achieved after treatment with doxorubicin, dacarbazine and cyclophosphamide combination chemotherapy. The other patient presented with the regression of lung metastasis, however the sacral mass progressed after treatment with doxorubicin and cisplatin.
Chou et al.6) reported the use of ifosfamide, epirubicin and cisplatin combination therapy. The sacral tumor progressed rapidly after chemotherapy and patient died of wound infection and uncontrolled sepsis one month later.
Casali et al.2) reported that imatinib mesylate has been found to have antitumor activity in patients with chordoma. This activity might be mediated by inactivation of platelet derived growth factor receptor β. Six patients with chordoma were treated with imatinib mesylate at a dose of 800mg daily for one year. Clinical and radiological improvement was seen in all patients.
In our study, we performed chemotherapy with 4 drug regimen including vincristine, ifosfamide, doxorubicin, etoposide, and following a single regimen of etoposide for the treatment of the systemic metastasis in the second case. After the chemotherapy, lung metastasis remained stable, however the tumors at the sacrum and mobile spine showed rapid progression. The patient presented chemotherapy induced bone marrow suppression and died of uncontrolled sepsis complicated by pancytopenia.
It is not certain whether chemotherapy is beneficial or which chemotherapeutic agent is effective in the treatment of metastatic DC yet. The combination chemotherapy based with doxorubicin, cisplatin and cyclophosphamide appears to be effective for some patients. However, physicians have to be alert using chemotherapeutic agents to manage associated complications, especially myelosuppression.
CONCLUSION
We experienced two cases of DC and reviewed the literature. DC is a rare and highly aggressive tumor. It is usually transformed from recurrent chordoma after surgical resection or RT. The treatment of choice is known to complete surgical removal of the tumor. In addition, there is short of evidence about the efficacy of adjuvant RT or chemotherapy. The prognosis is usually poor.