







INTRODUCTION
The functions of the skeleton are to provide mechanical support, to protect internal organs, and to provide a reservoir of calcium and phosphate for homeostasis of these ions. The compressive strength of bone, its ability to tolerate loads and its bending strength, the ability to resist bending during lever function, are achieved by the integrity of bone’s material properties and its structural design [1]. This review discusses those aspects of bone physiology that relate to certain new and potential treatments for bone loss, both inhibitors of bone resorption and anabolic pathways, especially through the parathyroid hormone (PTH) receptor pathway.
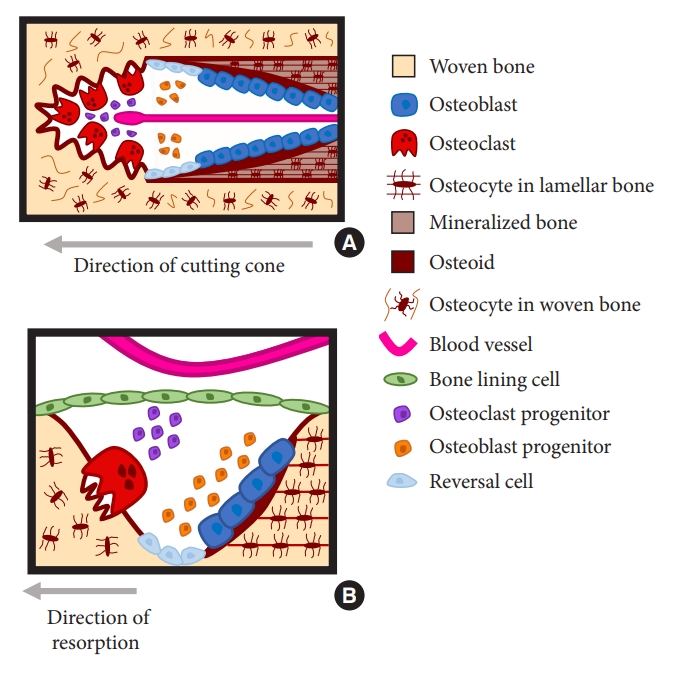
Like all structures, roads, buildings, bridges, bone undergoes fatigue damage, but bone is unique in its ability to repair itself, at least during young adulthood. This process of bone self-repair or bone renewal, is bone remodeling. The cells of bone are able to detect microdamage, and then replace either old or damaged bone with an equal volume of newly synthesized bone. Osteoclasts are responsible for removal of a volume of old or damaged bone. Osteoblasts synthesize new bone and to some extent dictate the volume new bone needed to refill the excavated cavity at a given location. Osteocytes, the most abundant cell of bone, are terminally differentiated osteoblasts that reside in lacunae throughout the skeleton and are interconnected with each other and with surface cells. These cells participate in the identification of the location and size of microcracks and, in part, orchestrate the resorption of the damaged bone and subsequent replacement with of a volume of new bone.
BONE REMODELING
An essential feature of bone physiology is that the cellular activities of bone cells ensure that the volumes of bone resorbed and formed at given locations throughout the skeleton are equal so that the total bone volume and its architectural design established during growth are preserved—at least during young adulthood. This process of bone renewal, or bone remodeling, takes place asynchronously where it is needed, in focal or discrete cellular packets throughout the skeleton, basic multicellular units (BMUs) separated from each other geographically and chronologically [2]. At some locations, BMUs are in their resorption phase, at other locations BMUs are in their formation phase, so that the sequence of bone resorption followed by bone formation by a BMU does not occur uniformly throughout the skeleton, an observation strongly suggesting that regulation of this cellular activity is locally, rather than systemically, regulated [3,4].
Resorption of a volume of bone by the osteoclasts of a BMU in human bone takes approximately 3 weeks while the bone formation by osteoblasts of that BMU takes longer, 3 to 4 months. At any time, the cellular activity of bone remodeling replaces only about 5%–10% of the skeleton annually, the remainder of the skeleton is quiescent. Renewal of the entire adult human skeleton takes about 10 years [5].
Understanding the tightly controlled processes of bone resorption and formation by individual BMUs throughout the skeleton requires appreciation of the many pathways that control cells of the osteoblast and osteoclast lineage and how they communicate with each other [6]. This process of bone remodeling is the result of biological activities produced by osteoclasts, osteoblasts and osteocytes as well as cytokines and growth factors produced by other cells, including those of the immune system and vasculature [6].
The first step in bone remodeling is the generation of active osteoclasts from hemopoietic precursors. One of the sources of osteoblast and osteoclast precursors for the BMU is via the circulation, through capillaries penetrating the canopy that overlies the BMU [7]. Osteoclast formation can take place rapidly in vivo, perhaps because there may be niches of partially differentiated cells available in the BMU [8], having arrived there in the circulation [9]. Regardless of the source of the initiation signal, osteoclasts are derived from early and late precursors available to sites that will become BMU’s, or recruited from blood available at the bone interface through a sinus structure of bone remodeling compartments identified and described in human bone [10,11].
At each of these sites, the resorption of a volume of bone is followed by new bone formation to refill the space. The BMU resorbs and replaces old bone at the same location so there is no change in bone size or shape. After a volume of bone is resorbed, the osteoclasts die or move away from the site. A reversal phase occurs in which a cement line is laid down. Osteoblasts then synthesize matrix, which mineralizes.
The cellular activity of bone remodeling occurs upon bone surfaces, the external or periosteal surface, and the 3 divisions of the inner or endosteal surface—the intracortical surface of the many canals traversing the cortex, the endocortical surface lining the medullary (marrow) cavity and the trabecular surfaces (Fig. 1). In the cortex, as reported initially by Hattner et al. [2], the osteoclast-mediated resorption takes place at points upon existing intracortical canals excavating a new canal by tunnelling through cortical bone, with subsequent ‘wave’ of bone formation refilling the newly formed osteon or bone structural unit with its newly formed central (Haversian or Volkmann) canal and concentric lamellae formed by mineralized collagen fibres. On endocortical and trabecular surfaces, pits or trenches are excavated forming hemiosteons under a canopy enclosing the process and penetrable by blood vessels [10].
An essential feature of bone remodeling is that just as the osteoblastic lineage cells control osteoclast formation, products arising from resorption of bone matrix and from the osteoclasts themselves, contribute to the promotion of osteoblast differentiation from precursors in the BMU, and hence to bone formation [3,5,7,12]. The formation of osteoclasts from hematopoietic precursors in the monocyte-macrophage lineage needs to be regulated through direct interaction with cells of the osteoblast lineage, stimulated by hormones, cytokines and prostanoids. Osteoblast lineage cells in bone together with immune cells in the marrow microenvironment play an essential role as a source of cytokines, including tumor necrosis factor α (TNF-α), interleukin (IL)-1 and IL-6, and PTH-related protein (PTHrP) [13].
Since the receptors for these and many other bone-resorbing paracrine factors are expressed in cells of the osteoblast lineage, they must first act on the osteoblast to modify osteoclast formation [14]. Despite the several different signaling mechanisms that these agents use upon their target cells, they converge to a common mechanism in promoting osteoclast formation, that requires receptor activator of nuclear factor kappa-Β ligand (RANKL) as the essential mediator of osteoclast formation in response to all known stimuli [15]. The osteoblast lineage also produces the decoy receptor, osteoprotegerin (OPG), a soluble member of the TNF receptor superfamily, which is a very effective inhibitor of osteoclast formation that acts locally as a “brake” on osteoclast formation [16]. Osteoblasts/stromal cells are also the source of macrophage colony-stimulating factor, which plays a crucial role in osteoclast formation by promoting the proliferation of precursors.
While bone remodeling remains balanced during young adulthood, around midlife in both sexes, there is a reduction in the volume of bone deposited by each remodeling event so that remodeling becomes unbalanced. Each remodeling event deposits a smaller volume of bone than was resorbed resulting in net negative remodeling imbalance producing a reduction in bone mass and focal microarchitectural deterioration; cortices thin and become porous, trabeculae thin and perforate.
In women, menopause results in estrogen deficiency, the birth rate of remodeling units increases and there is worsening of the remodeling imbalance—the volume of bone resorbed by each remodeling event increases and the volume of bone deposited decreases further than produced by advancing age alone. Bone loss accelerates and microarchitectural deterioration worsens predisposing to fractures [17]. This remodeling imbalance is the necessary and sufficient cause of bone loss and identifies 2 targets from prevention of the skeletal deterioration—the use of antiresorptive agents to reduce or prevent bone resorption and the use of anabolic agents to stimulate and increase bone formation.
ANTIRESORPTIVES
As understanding of bone biology increased, new insights guided the development of antiresorptive therapies for osteoporosis. The clinical outcomes of these new therapies could be predicted because the actions of the selected cellular target, osteoblasts, osteoclasts and osteocytes were known, and in some cases preclinical evidence fulfilled those predictions. The aims were to develop therapies to improve fracture risk reduction, and in doing so, to avoid long term deleterious effects on bone material composition and structure.
The antiresorptive era in the treatment/prevention of bone fragility began with the first successful double-blind, placebo-controlled trial, which was that of alendronate in the mid-1990’s [18]. This was followed by the successful establishment of several other bisphosphonates, and finding that zoledronate is extremely potent and long-lived in its actions, so that annual infusion, or even a single infusion reduces the risk of fractures and slows bone loss in women with osteoporosis or more modest deficits in bone mass (osteopenia)[19], the latter being a most important observation because most women having fragility fracture have osteopenia, not osteoporosis [20].
Bisphosphonates are the most common treatments used clinically to prevent or partly reverse bone fragility but increasingly, denosumab is used because of its powerful remodeling suppressant action. Studies establishing the essential physiological roles of RANKL and OPG in controlling osteoclast formation and activity, revealed a pathway rich in targets for pharmaceutical development. Denosumab is a fully human monoclonal antibody against RANKL, that binds with high affinity and specificity to RANKL to inhibit its action. It has an exceptionally prolonged and powerful action and in therapeutic regimes is administered by subcutaneous injection every 6 months [21]. Denosumab virtually abolishes the birth rate of BMUs by preventing RANKL from promoting osteoclast formation and by promoting the apoptotic death of existing osteoclasts already resorbing bone [22]. The phase III study using subcutaneous injection of denosumab every 6 months resulted in substantial reduction of vertebral, nonvertebral and hip fractures, and striking suppression of bone remodeling that was more marked than achieved by bisphosphonates [21], drugs that reduce but do not abolish bone remodeling [17].
Discontinuation of denosumab treatment is associated with a rapid restoration of remodeling with overshoot of remodeling markers above the baseline pre-treatment levels [23], rapid bone loss and a reduction in bone mineral density (BMD) to baseline, but usually not below baseline. Of concern is the occurrence of multiple vertebral fractures in 4.2 per 100 patient-years compared to 3.2 in the placebo group. Of the 56 participants with new vertebral fractures, 60.7% had 2 or more and 23.2% had 4 or more fractures compared to multiple fractures in 38.7% of participants given placebo [24].
In the clinical trials that established the efficacy of these antiresorptive drugs, the major endpoint has been the occurrence of symptomatic and asymptomatic vertebral fracture identified by lateral spine radiographs [25]. While vertebral fractures are the flagship of fragility fractures signaling the presence of bone fragility, epidemiological studies show that 80% of all fragility fractures are nonvertebral (distal radius, proximal humerus, pelvic, hip) [26], and hence the need for therapies shown to reduce the risk of nonvertebral and hip fractures as well as vertebral fractures. Nevertheless, vertebral fractures are associated with significant disability [27] and reduced survival [28]. Moreover, vertebral fractures, like all fragility fractures, increase as age advances and a substantial proportion are asymptomatic and so remain underdiagnosed. These fractures can cause spine deformity (dowagers hump, kyphosis), chronic back pain, impaired respiratory function, pain due to impingement of the lower ribs upon the pelvis. Importantly, vertebral fractures increase the risk of further vertebral and nonvertebral fractures [29].
ANABOLIC AGENTS
1. PTH as an Anabolic
Antiresorptive agents like denosumab abolish bone remodeling and so prevent bone loss, further deterioration in bone mass and microarchitecture. Bisphosphonates slow bone remodeling; they do not eliminate bone loss [30] and so these agents slowed but did not abolish bone loss and microarchitectural deterioration —the deficit in bone mass and microarchitectural deterioration at the time of presentation is not restored by antiresorptives of any kind. The increase in BMD is largely the result of a reduction in the transitory ‘remodeling space’ deficit in bone matrix produced by a reduction in the number of resorption cavities excavated, partial refilling of resorption sites excavated before treatment and completion of mineralization of matrix resident in bone for a long enough period of time in the face of reduced bone remodeling [17] so that the slow process of completion of mineralization can take place.
Until recently, no therapeutic approach was available to restore compromised bone mass and microarchitecture. That situation changed with the development of PTH as an anabolic therapy for the skeleton, despite its better-known action as a resorptive hormone, and also the development of neutralizing antibody against sclerostin, the osteocyte-derived protein that is a powerful inhibitor of bone formation [31-33]. As a result of the advances made in understanding of PTH, the approved therapies in several countries are PTH (1–34) and PTH (1–84), and more recently abaloparatide [34], which is structurally related to both PTHrP and PTH (vide infra).
WHAT IS PTHrP?
PTHrP was discovered during studies of the syndrome of humoral hypercalcemia of malignancy in which the biochemical features parallel closely those of PTH excess (primary hyperparathyroidism). This was reflected in the fact that for many years the syndrome had been considered to be due to “ectopic” PTH production by those cancers [35], until in the 1970’s, when immunological methods revealed that the cancer-derived activity, although it mimicked PTH actions, differed from PTH immunochemically and must therefore differ in structure [36,37].
Cloning and sequence analysis revealed PTHrP as the cause of the humoral hypercalcemia of malignancy, and to be closely structurally related to PTH, in that 8 of the first 13 residues in each molecule are identical. Beyond that, there are no more identities between PTH and PTHrP than would be expected by chance. This sequence homology in the first 13 amino acids is sufficient to explain why these molecules act equally through the same G protein-coupled receptor PTH 1 receptor (PTH1R). Despite the close homology, it nevertheless did not prove difficult to generate antibodies to the amino-terminal domains that distinguish between the two. This became evident from the first antipeptide antibodies produced [38]. Furthermore, consistent with the activity being restricted to the N-terminus, PTHrP recombinant proteins of 84, 108, and 141 residues are all equipotent on a molar basis with shorter amino-terminal peptides of either PTH or PTHrP in activating adenylyl cyclase in target cells [39,40]. In addition, abaloparatide, which is also based on the N-terminus, is equipotent with teriparatide in the same biologiocal assay [41,42].
PTHrP cannot be detected convincingly in postnatal plasma in healthy subjects using assays based on the N-terminal domain [43,44], a major contrast with PTH, which is a hormone secreted only by the parathyroid glands. Evidence accumulated rapidly that PTHrP is produced in many normal tissues including keratinocytes, endothelial cells, bone, smooth muscle cells, lactating mammary tissue, and neuroendocrine tissues including pancreas. In these tissues, PTHrP was found to have multiple actions, including relaxation of vascular and other smooth muscle beds, including gastrointestinal and uterine, as well as promoting transplacental calcium transport, and stimulating bone remodeling [45-47]. These findings indicated that PTHrP, unlike PTH, functions as a paracrine regulator of cell function. The only non–tumor-associated circumstances in which PTHrP is found in the circulation and can therefore act as a hormone, are in the fetus and in the maternal circulation during lactation. Indeed, the role of PTHrP in bone remodeling is a local one, and the use of PTH pharmacologically is likely to make use of the local physiological function of PTHrP as a promoter of bone formation [47].
Revelations provided by the structure of PTHrP pointed to new ways of presentation to the PTH receptor, given the equal potencies of PTH (1–34) and PTHrP (1–34) in promoting cyclic AMP (cAMP) production in target cells. From the first in vivo experiments though [48], it was evident that the anabolic efficacy of PTHrP was much less than that of PTH (1–34). Such a lesser potency in vivo could be anticipated because even these truncated forms of PTHrP have many target sequences that are susceptible to proteolysis [49]. The same applied to studies in human subjects. The daily dose of PTHrP (1–36) in human subjects required to increase bone formation markers was manyfold higher than the dose of teriparatide needed to achieve these marker levels [50], likely due to the proteolytic cleavage described above. The higher dose requirements might thus be due to a difference in pharmacokinetics, with PTHrP (1–36) degraded more rapidly following injection, and so not as widely distributed to activate BMUs. The result would be that lesser amounts of active agonist would be available to the receptor.
Early studies by Stewart and colleagues suggested that PTHrP (1–36) action was relatively free of a resorptive effect as determined by measuring serum biochemical markers of bone remodeling. Similar claims have been made for abaloparatide [34,51], which is a 34-amino acid peptide in which the first 21 residues are identical with PTHrP. Abaloparatide also contains a number of substitutions that were planned to improve stability [41,52], though the most susceptible of the PTHrP cleavage sites (–R19R20R21-) is located within both PTHrP (1–36) and abaloparatide. Abaloparatide (80 μg per day) and teriparatide (20 μg per day) have been reported to have comparable antivertebral fracture efficacy, with claims that this is achieved with a lesser effect on bone resorption markers with abaloparatide than with teriparatide [34,51]. However, in the same data, the formation marker level was also less with abaloparatide, so the claim of a lesser effect on resorption is not well founded. There remains no information on the cellular basis by which PTHrP (1–36) or abaloparatide could have a lesser effect on bone resorption than formation, so abaloparatide remains as a peptide analogue with effects similar to those of teriparatide.
MECHANISMS IN THE ANABOLIC EFFECT OF PTH
Although early concepts of the pathogenesis of osteoporosis related estrogen deficiency to postmenopausal bone loss, rapid advances were achieved with the development of ovariectomized (OVX) rat and mouse models of bone loss. These arose from the work of Kalu [53] and were made use of in many studies of the anabolic action on bone of daily PTH injections [48,54].
These unequivocal demonstrations of the anabolic action of PTH had come unexpectedly, since it was widely believed that PTH acted strictly as a hormone that regulates extracellular calcium levels. This had been accepted since the mid-1920’s, when the first biologically active extracts of parathyroid tissue were found to raise blood calcium when injected into animals [55]. It had required the development as much as 50 years later, of such methods of demonstrating an anabolic action of PTH, using the new OVX rodent response, to re-establish interest in the findings of Albright et al. [56] and Selye [57]. What they had found was that daily injection of PTH (in those days a tissue extract) resulted in an increase in the amount of bone in rats.
It provides an interesting comment on the rate of progress of medical research in that era, that further work on the anabolic action of PTH took such a long time to follow. Much research interest in PTH was generated following the purification, sequence and cloning of PTH and the finding that the amino-terminal domain appeared to have the full activity of the hormone (reviewed in [55]). This led to a limited clinical study of administration of PTH (1–34) to postmenopausal women, in which circulating marker effects appeared consistent with an anabolic response [58].
That clinical study [58] used a PTH (1–34) dose that was calculated as low, but it still appeared that any excess of injected PTH was accompanied by a resorptive effect. This was noted regularly in the experimental studies that followed, in that PTH treatment was often accompanied by a resorptive response, featured by hypercalcemia that resulted from increased osteoclast formation and bone resorption [59]. Such a resorptive response could be enhanced when PTH levels were maintained high by infusion in vivo over a period of time [60].
From the clinical therapeutic point of view the major advance was the double-blind, placebo-controlled clinical trial of PTH (1–34) (teriparatide), showing declines of vertebral fractures of 65% and nonvertebral fractures by 53% in women with postmenopausal osteoporosis [61]. This led to acceptance of teriparatide by the U.S. Food and Drug Administration, and to their approval of peptides acting through the PTH receptor, including PTH (1–84) and a new analogue abaloparatide, that are accepted treatments in some countries.
The difference between pharmacologic effects of intermittent and sustained treatment with PTH was demonstrated in a study in rats in which it was shown that when PTH was administered either by intermittent subcutaneous injection or infusion, but using the same daily dose for each method, intermittent injection yielded an anabolic effect, whereas continuous infusion resulted in skeletal loss due to osteoclastic resorption [59]. Other work showed that subcutaneous injection of PTH resulted in a transient stimulation of expression in osteoblasts of RANKL, whereas maintaining PTH serum levels elevated over 4 hours resulted in persistence of the increased RANKL expression, with the implied increased production of active osteoclasts and resorption [60].
Direct information on the action of intermittent PTH therapy on human bone is very limited in comparison with the experimental data, because of the difficulty in obtaining human biopsy material before and after treatment. The human material available is derived by biopsy from the iliac crest, which is not a weightbearing bone and therefore does not show the bone-building effect of weightbearing. Having said that, those studies in which biopsy material has been studied by histomorphometry after PTH treatment have agreed that the anabolic effect is the result predominantly of stimulating remodeling by increasing the number and activity of BMUs, with a net increase. In the amount of bone at each BMU [62-65]. There is some effect on modeling also, some of this related to what is described as “overfilling” of BMUs, where bone is formed on surfaces extending beyond the resorption pit [63,66]. Making a distinction between remodeling and modeling effects of PTH anabolic treatment might become important when considering use of PTH in subjects treated with maximally effective resorption inhibitors, where the osteoclast formation and activity essential for remodeling are prevented.
Thus any effect of PTH on modeling provides an attractive way of making use of the direct action of PTH on the osteoblast lineage, where the chief actions relevant to a modeling effect are the promotion of differentiation of committed osteoblast precursors [67], inhibition of osteoblast apoptosis [68], and action upon osteocytes to inhibit the production of sclerostin, a powerful inhibitor of bone formation [69].
With the use of PTH injection to promote bone formation, the blood levels of formation markers (e.g., P1NP [procollagen type I N-terminal propeptide]) are rapidly increased at the outset, and progressively increase thereafter [61], whereas any increase in bone resorption markers (e.g., CTX [C-terminal telopeptide of type I collagen]) begins to take place only several weeks later. The likely explanation of this is that the formation marker response represents an acute cellular response to PTH; the matrix accumulated after several weeks would constantly be resorbed as a result of osteoclast activity, and would eventually reach a level that would be detectable in serum and/or urine.
When it was recognised that PTHrP is the paracrine ligand for PTH1R in bone [70,71], several truncated forms of PTHrP that include the PTH1R—interacting portion of the molecule have been investigated as anabolic agents. These include PTHrP (1–34), PTHrP (1–36), and PTHrP (1–74). The anabolic action of PTHrP (1–36) in human subjects, as assessed by measurement of bone formation markers, has been suggested to be relatively free of the resorptive action that is an essential component of the remodeling action of PTH [50,72,73]. In these studies of PTHrP (1–36) daily injection, the doses required to increase levels of anabolic markers are many-fold higher than those of PTH. The higher dose requirement might reflect a pharmacokinetic difference because of the greater proteolytic susceptibility of PTHrP, as discussed earlier in the review.
PTH AND PTHrP – DIFFERENCES IN RECEPTOR INTERACTIONS
Another explanation has been offered for the different action of amino-terminal PTHrP, based on differing interactions with PTH1R of the amino-terminal domains of PTH and PTHrP. In studies of initial interactions at the plasma membrane, the action of PTHrP (1–36) was found to be restricted to the cell surface, while PTH (1–34) was internalized and brought about a more prolonged increase in cAMP in the target cells [74]. The authors suggested that such a differential response might explain why daily injection of PTH (1–34) elicits a resorption response, while PTHrP (1–36) does so to a much lesser extent. The same argument has been applied to the new analogue, abaloparatide, which is identical to PTHrP in its first 21 residues, but has 8 residues different from PTHrP between 22 and 34 [52], and which behaves in a similar manner to PTHrP (1–36) in its initial interaction with PTH1R [41]. Abaloparatide has also been suggested to have a lesser effect on resorption markers than teriparatide in patients [51], although the 2 peptides have indistinguishable effects on bone in mice [75]. These reported differences in internalization and cAMP formation [41] are of some interest, but extrapolating them to explain in vivo effects is questionable without any evidence beyond cAMP generation, and without undisputed differences in clinical data between formation and resorption outcomes.
COMBINATION THERAPY – ANTIRESORPTIVES WITH INTERMITTENT PTH
A question that has commonly arisen in discussing PTH anabolic action is whether there is anything to be gained by treating with PTH combined with an antiresorption drug? Quite the reverse was suggested in an early study that found the anabolic effect of PTH was significantly reduced in sheep cotreated with the bisphosphonate, tiludronate [76]. Some, but not all studies in rats, as well as some clinical studies [77,78] have also shown impaired anabolic responses with such combined treatments. The advent of the even more powerful resorption inhibitor, denosumab, led to further studies of combined therapy [30]. An additive effect on BMD has been observed in patients receiving high dose PTH that was then supplemented with denosumab [79-81].
However, a deficiency of these studies is the use of BMD as the endpoint, which should not be taken as a measure of bone mass [30]. No real outcomes on bone mass, or antifracture efficacy have yet been reported in the denosumab/teriparatide combination studies. Ultimately, studies of fracture incidence are needed, but they present such difficulties that it is not easy to see them being undertaken. The question remains therefore, and presents an attractive proposition. If PTH combined with virtually complete osteoclast blockade (using denosumab) does indeed result in a useful anabolic effect, it points to an osteoclastfree anabolic effect of PTH, that is, an effect through promotion of modeling of bone.
It is appropriate to emphasise in this context, that the generation and resorptive activity of osteoclasts is an essential feature of bone remodeling, and inseparable from the process [6,82]. Thus the distribution of PTH anabolic effect between remodeling and modeling is an important matter that impinges upon any therapeutic changes that might require predominant actions through one or the other mechanism. Anabolic action using the PTH pathway in the absence of osteoclasts surely would use the direct action of PTH on the osteoblast lineage, which is well established. It would then use either a stimulation of bone modeling, or direct action upon osteoblastic cells that were within BMUs when osteoclast formation and activity was blocked, and perhaps also inhibition of sclerostin production by osteocytes.
Thus, in the absence of osteoclasts, PTH can still exert an anabolic action to some extent, although it is not yet clear how exactly it would do so. It could possibly be a direct action on osteoblast lineage cells within BMU’s that were existing at the time of RANKL blockade, through a direct modeling action of PTH, and/or PTH inhibition of sclerostin production by osteocytes [13,66,69]. Whether such combination treatment approaches will have favourable effects on structure or on fracture incidence will be a difficult question to answer, largely because there will be great reluctance to undertake the large clinical studies needed to assess fracture risk.
WNT SIGNALING TARGETS – SCLEROSTIN
Research of the last few decades has led to a focus on activation of the canonical Wnt signaling pathway in promoting of formation, providing another prospect in anabolic therapy for the skeleton. The link between Wnt signaling and human bone disease came from studies in rare monogenic disorders, the osteoporosis, the osteoporosis-pseudoglioma syndrome (OPPG, OMIM 259770) caused by loss-of-function of LRP-5 and charecterised by severely decreased bone mass, and a syndrome of high bone mass found to be caused by a gain-of-function mutation of LRP5 (OMIM 601884). The Wnt/β-catenin signaling pathway has a number of inhibitors and activators that offered several targets suitable for pharmacological intervention [32,33,83], aiming to increase Wnt/β-catenin canonical signaling in order to increase bone mass. Sclerostin, the protein product of the Sost gene, is produced primarily by osteocytes and powerfully inhibits bone formation through inhibition of Wnt signaling, sparking great interest in roles for the osteocyte in bone modeling and remodeling. Sclerostin null mice have very high bone mass, and conversely, severe osteopenia occurs in transgenic mice overexpressing sclerostin in osteocytes [84]. Loss of function Sost mutations cause the greatly increased bone mass of sclerosteosis and van Buchem disease [85]. Physiologically, rapid reductions in sclerostin could mediate the reduction in refilling of an excavated resorption site so that it does not overfill [31]. Initial success in animal models was reported with the inhibition of DKK1 (dickkopf related protein 1), GSK-3 (glycogen synthase kinase 3), and sclerostin [33,86,87], but the most advanced in clinical development is blockade of the action of sclerostin by treatment with a neutralizing antibody. A 12-month randomized, placebo-controlled, multidose study of 410 women was carried out with the humanised monoclonal antibody (Romosozumab) recapitulated the rapid increase in BMD seen in the preclinical studies [88]. In the Romosuzomab trial there was a statistically significant increase in cardiovascular events, requiring a limitation on Romosuzomab use.
In discussing the PTH anabolic action we drew attention to the fact that cessation of treatment is followed by bone loss and increased resorption markers. The same occurs in the case of Romosuzomab treatment also, and the question of using that anabolic therapy concurrently with an antiresorptive has been discussed. In a study of sequential alendronate and antisclerostin in OVX rats, this cotreatment did not blunt the anabolic effects of anti-sclerostin on bone formation. In clinical studies, BMD increases were greater when Romosuzomab was used before antiresorptive treatment with denosumab [89].
FRACTURE RISK REDUCTION – WHY, WHO, WHEN, WHAT DRUG?
Fragility fractures are a public health burden because they occur in 1 in 3 women and 1 in 5 men. Fractures increase morbidity, mortality and cost to the community [90]. Fractures are the result of bone fragility produced by age-related deterioration in bone remodeling which compromises bone’s material composition and microarchitecture and increasing risk of falling [1].
Identification of persons at risk before the first fracture is achieved by measurement of BMD. This approach identifies those at high risk with ‘osteoporosis’ (BMD T score ≤ -2.5 standard deviation), but over 70 percent of women and men having fragility fractures have the modest BMD deficits of osteopenia (BMD T score: -1 to -2.5 SD) [19,20]. The risk of fracture in these individuals is due to cortical and trabecular microarchitectural deterioration. Identification of individuals is achieved using high resolution computed tomography, a new noninvasive imaging method that has now become available [91]. Assessment of clinical risk factors using a fracture risk assessment score (FRAX) [92], trabecular bone score [93] and the use of biochemical markers of bone remodeling to identify persons losing bone rapidly, also assist in risk assessment [94].
The choice of therapy depends on the quality of evidence of antifracture efficacy against all 3 major classes of fracture—vertebral, hip and nonvertebral fracture. Only 4 antiresorptive drugs are proven to reduce these fractures—risedronate, zoledronic acid, denosumab, and hormone replacement therapy. The drugs reduce the risk of vertebral and hip fractures by 50%–60% and nonvertebral fractures by 30%–40%. In the fracture intervention trial, alendronate reduced vertebral and hip fractures but not nonvertebral fractures. Whether one antiresorptive has greater antifracture efficacy than the others is not known. Any of these 3 drugs are suitable first line approaches to treatment with choices modified according to potential adverse events and patient preference. The antifracture efficacy of calcium supplements, vitamin D alone or with calcium has not been demonstrated [95].
Treatment is long term, and probably needs to be permanent (like treatment of hypertension and cardiovascular disease). One possible exception is the use of bisphosphonates because these drugs are adsorbed into bone matrix remain resident in matrix for many years. When stopped, bisphosphonates are resorbed from bone as remodeling continues, some drug is excreted in the urine or readsorbed into bone matrix and continues to act so that a drug ‘holiday’ is feasible because treatment is effectively continuing. If remodeling and bone loss recur, treatment should be restarted. Cessation of denosumab is challenging for reasons explained above. If cessation is desired, weaning should be slow with replacement by another antiresorptive therapy. How this is to be done is still a matter of contention. The concern about osteonecrosis of the jaw or brittle bone disease is often a basis for stopping therapy but these adverse events are uncommon and occur with both bisphosphonates and denosumab (but not with weak antiresorptives like calcium supplements and raloxifene).
Anabolic agents are usually reserved for patients with prevalent fractures or evidence of severe bone loss and microarchitectural deterioration. However, if anabolic therapy has better antifracture efficacy than antiresorptive therapy then a case can be made for anabolic therapy as first line treatment, a currently ‘fashionable’ approach that warrants close interrogation. Teriparatide, abaloparatide, and romososumab have all been convincingly shown to reduce the risk of vertebral fractures and there is indeed evidence for greater antivertebral fracture efficacy of teriparatide over risedronate [96] and romososumab over alendronate [17].
However, 80% or more of all fractures are nonvertebral (forearm, proximal humerus, ankle, rib, pelvis) or hip. Evidence for anti-hip fracture efficacy is not available for teriparatide relative to placebo, perhaps because the pivotal trial was stopped prematurely due to concerns about the occurrence of osteosarcomas in rats. Nor is there evidence for anti-hip fracture efficacy of abaloparatide or romosozumab. There was evidence for anti-hip fracture efficacy of romosozumab when followed by alendronate for 2 years. Antinonvertebral fracture efficacy has been reported using teriparatide (but only in one of several studies), not for abaloparatide and not for romosozumab except when followed by alendronate or denosumab. Absence of evidence is not evidence of absence of course. These trials were brief and sample sizes may have been insufficient to demonstrate superiority of one effective agent over another. Whatever the case, if anabolic therapy is to be used, it must be followed by antiresorptive therapy for the benefits in BMD and microstructure to be sustained.










")













